我2017年毕业以后进入头部基因公司医学信息中心做生信,前半年主要做NGS数据质控、RNA-seq、WGS和WES等比较传统的生信分析。2018年开始负责临床病原微生物基因检测产品的生信分析,也就是临床mNGS。 mNGS在临床上推广也是从那时刚刚开始,一个新人进入了一个新的领域,也算得上是最早的那批人了。2019年到现在的公司后至今还是一直在做mNGS,在mNGS领域摸爬滚打了近7年,应该算差不多可以入门了,也只会做这个。 虽然时至今日已经有很多关于mNGS性能评估及应用的文献或专家共识发表,但从跟很多医生或专家的交流中感受到,大部分人对mNGS的生信内容还是很模糊的认识。基因组数据库怎么来的,物种分类怎么做,背景菌怎么分析,假阳假阴怎么回事,分析结果准不准等等都是一些常见的问题。很多医生,包括监管部门都不会只满足于“科普型”的解释,都很关心这些问题的真正难点是什么?能不能解决?有没有具体的解决方案等等。而这一些问题都是少有甚至没有文献可以参考的。 这些问题总结起来就是:mNGS生信面临的真正挑战是什么? 这是一个非常宏大的问题,此外,临床mNGS完整流程包括:样本采集、运输、湿实验、干实验(生信)、临床解读等等一系列环节,每个环节都有非常深入的学问,并且环环相扣。
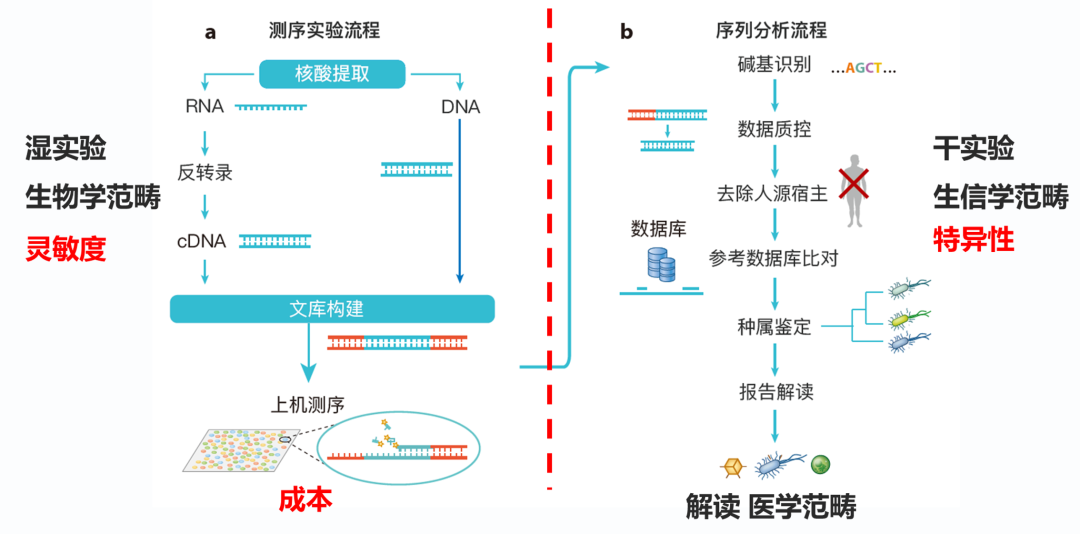
图1. mNGS全流程示意 生信其实跟每一个环节都关联,脱离了任何一个环节的信息都可能无法完成分析或者得出准确的结果。但限于能力和篇幅,这里仅聚焦于讨论纯生信的部分。
图2. mNGS湿实验和干实验的内容。湿实验属于生物学范畴,难点是要解决灵敏度的问题,样本里面有什么就能测出来什么;干实验是信息学范畴,难点是要解决特异性的问题,测序数据里面有什么就要准确鉴定出来什么,同时还要能区分其来源(来源于样本还是实验噪音)。 这些年无论是给内部培训,还是给外部分享,多多少少都有分享一些经验或看法。如今梳理总结一下,纯个人鄙见,不全面,也不一定对,欢迎交流。 要回答这个问题,我们得先从mNGS的临床需求入手。临床的需求是什么?mNGS可以解决临床的这些需求吗? 从应用场景的角度看,mNGS主要应用于疑难、急危重症的感染患者。 “疑难”说明可能常规的检验方法学都试过了,没有查出真正病原,所以对mNGS的需求就是具备“全面且准确”的检测能力,能覆盖传统方法学以外的检测范围或灵敏度。“急危重症”,说明时间等不起,需要尽快明确病原体马上对症下药,所以对mNGS的需求除了“全面且准确”外,还有“快”的需求。还有一点,从患者的角度考虑,还需要做到“经济可负担”。 “全面、准确、快、经济可负担”这些可能就是mNGS的核心问题。需要再次强调,mNGS是一个整体的技术,解决这些核心问题也是需要从全流程去考虑的,生信只是其中的一部分。如果将这些问题拆解到生信里面去,对应的几个关键点就是“基因组数据库全面(决定着检测范围)、物种分类鉴定准确、分析时间快、硬件成本可负担”等。 这里面的任何一点单独做到也许是简单的,但是要全部满足似乎就是一个“不可能三角”。基因组数据库做“全”了也许会因为基因组错误的积累或者同源干扰太多导至假阳假阴增加,无法做到“准”,也因为数据库太大导至分析时间长而无法做到“快”,或者靠堆硬件计算资源实现了“快”而导至“成本”很高。兼顾所有是mNGS生信的真正挑战所在。 先看看mNGS生信分析流程的一个例子:
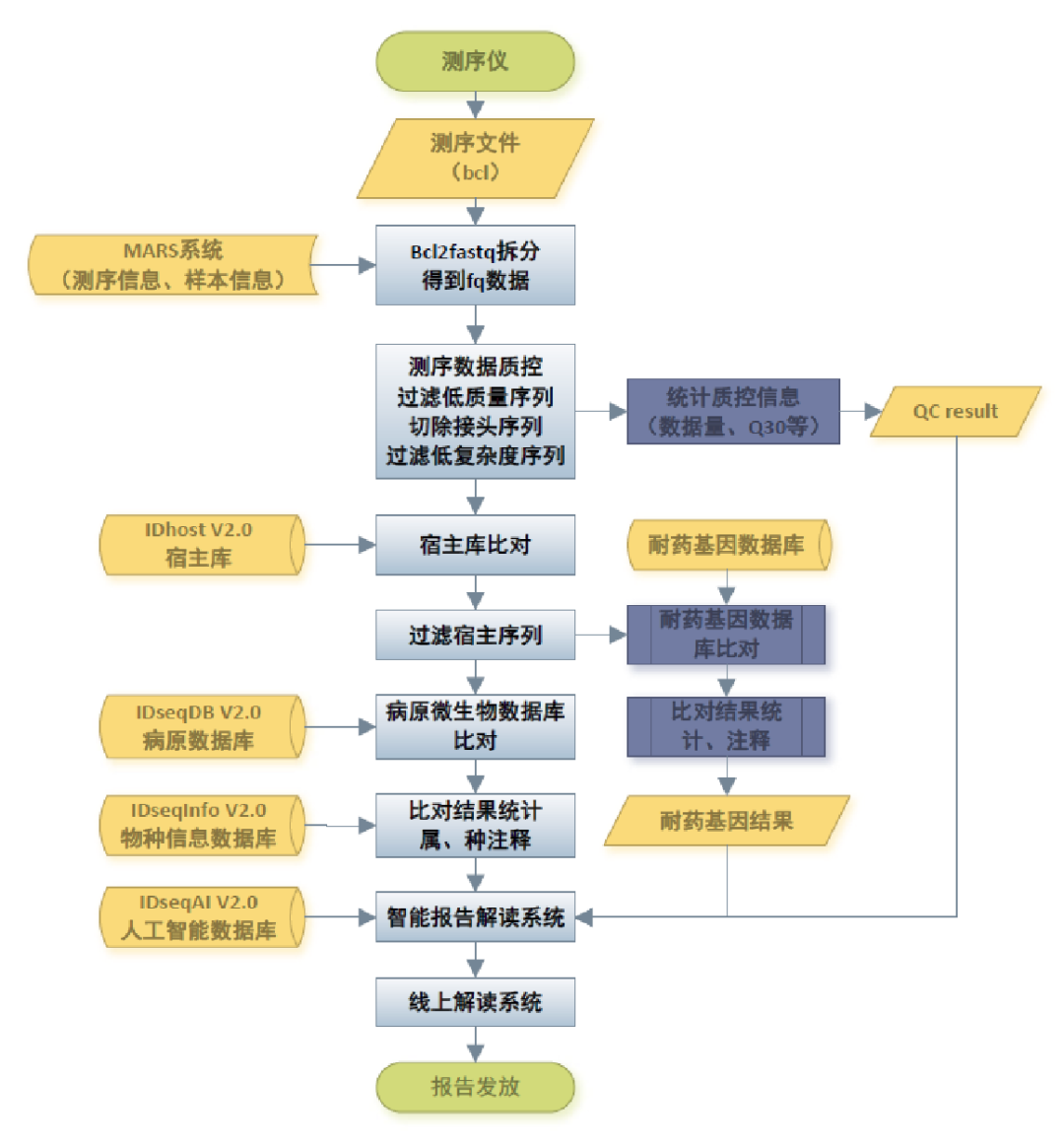
图3. mNGS生信分析过程。主要过程为:测序>fastq拆分>数据质控>人源序列过滤>微生物数据库比对>种属分类鉴定>报告解读等。有些还可能包含耐药和毒力的分析过程。 人源序列过滤、微生物基因组数据库、分类鉴定算法、报告解读等这些内容是影响mNGS生信性能的关键所在。 人源序列过滤是保证mNGS结果准确的关键之一 因为标本是从人身上采集的,测序数据里面大部分序列都是人源的,可达95%以上,若湿实验环节没有去宿主措施甚至可达99%以上。如果不对这些人源序列进行过滤,在微生物数据库比对环节,一是因为数据量太大所需的比对时间特别长,二是因为基因组同源或者微生物污染人源序列的问题,导至很多微生物的假阳性检出。 人源序列过滤的挑战在于,不仅要过滤,还要过滤得“全”。为什么要过滤得“全”?因为微生物基因组污染了大量的人源DNA,至于这些污染是怎么来的可以参考图4及其文献。
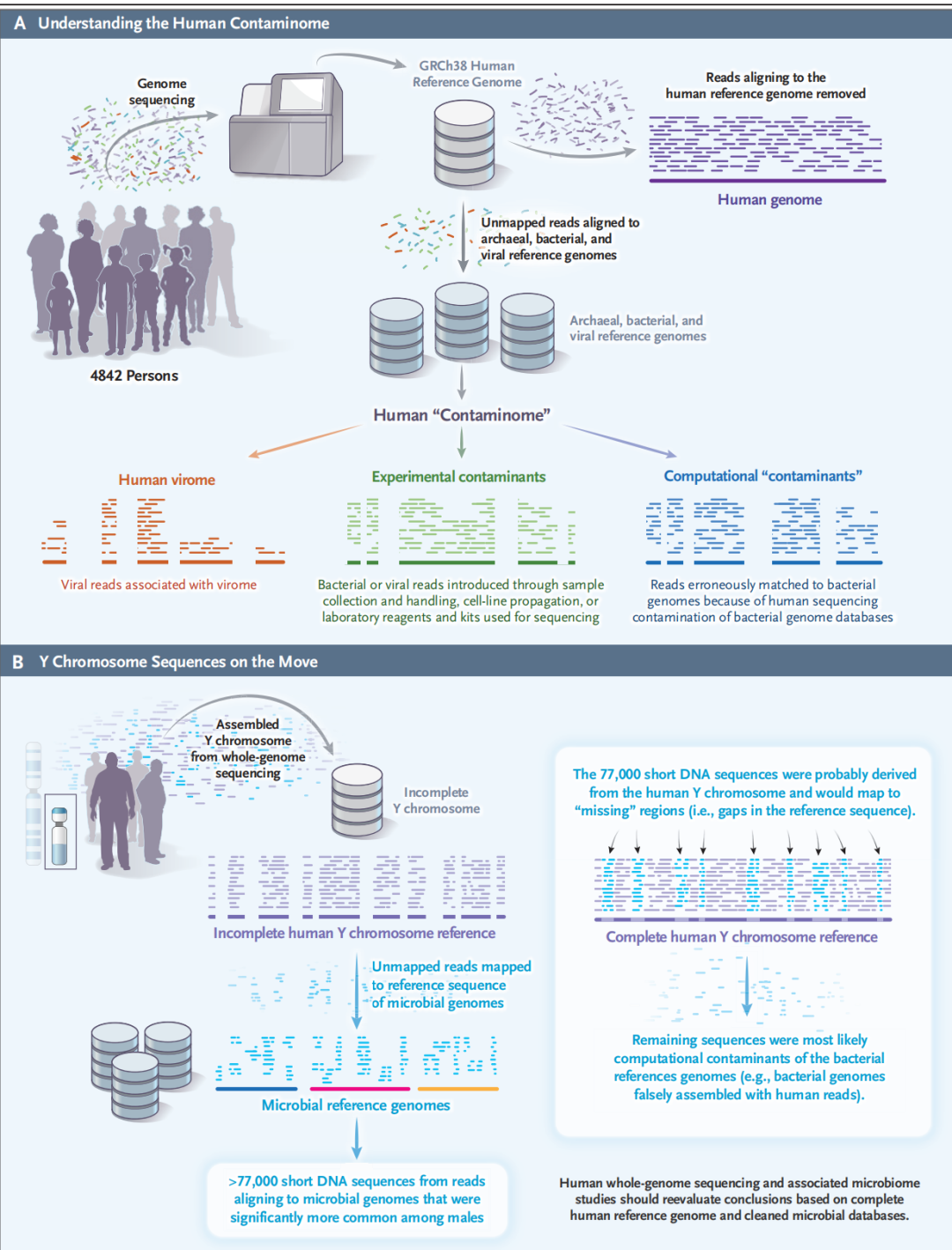
图4. The Human “Contaminome” and Understanding Infectious Disease. Simner PJ, et al. N Engl J Med. 2022 人类参考基因组不管是GRCh38,还是刚发布不久目前最完整的T2T-CHM13,或者黄种人的YH2.0,其实都还是个人基因组或者少数几个人的基因组。英国15万人群基因组测序项目的分析表明平均每个个体基因组存在约341万个SNPs或indels,且不同地域(欧洲、非洲、亚洲)人群间的变异差异更显著(https://doi.org/10.1038/s41586-022-04965-x)。对于人类来说,这个变异的比例也许很小,但是在mNGS测序数据里面,如果宿主序列占比为99%,人类序列过滤后即使只有1%的残留,在剩余的序列里面,人类的序列占比仍然高达约50%!这些序列在后续流程中都是要进行微生物数据库比对的,会产生多少假阳性? 我相信做过mNGS生信或解读的人在这点上是有体会的:明明已经过滤了人源序列,为什么检出的病原体序列拿去BLAST比对是时候还是会比上人的序列?
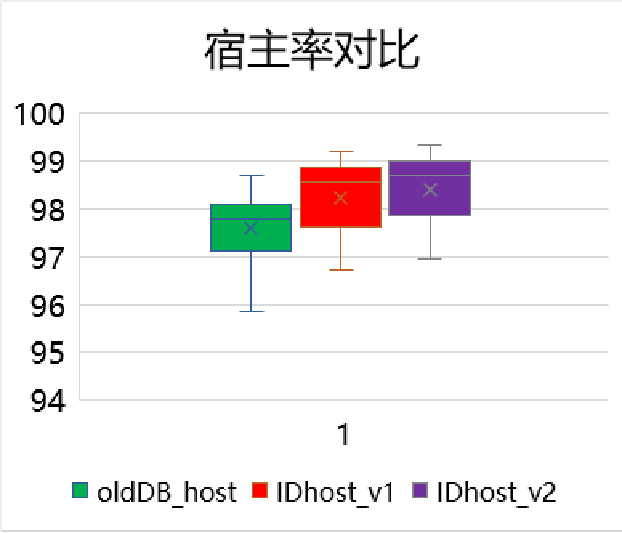
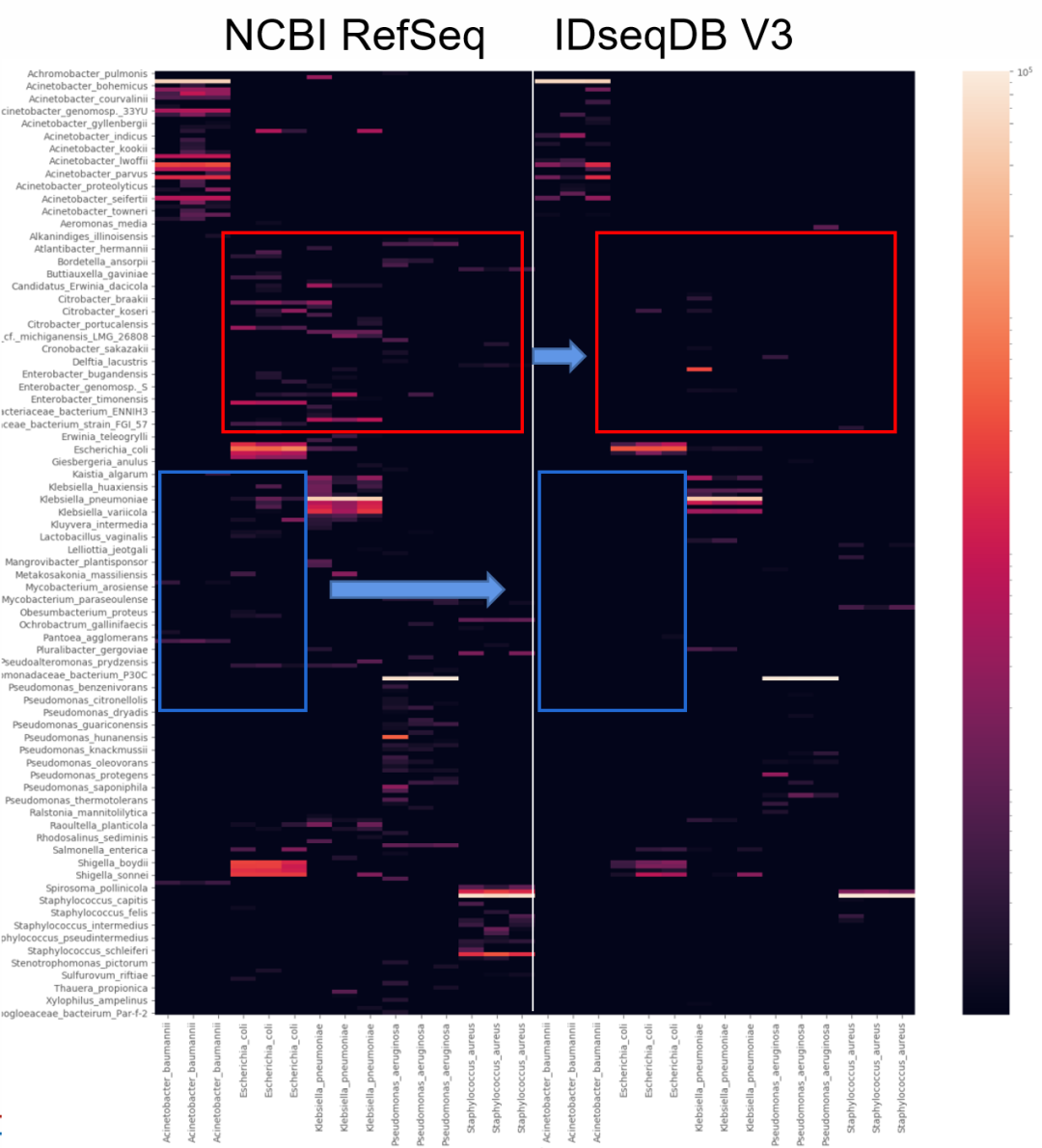
图5. 一例样本检出了Trichosporon asahii(阿萨希丝孢酵母),提取其序列进行在线BLAST比对,结果却比对上人类序列。 所以解决的方案就是需要构建一个“人类泛基因组”数据库用于过滤人源序列,可综合GRCh38、T2T-CHM13、YH2.0和其他人类参考基因组和其RNA序列,以及尽可能收集NT库里面人源相关的序列。由于人线粒体DNA的多态性非常高,因此也需要将人线粒体DNA数据库加入到人源数据库中。 图6是人源数据库三个版本升级过滤人源序列的效果对比。对于已经测序的数据,分析的宿主率越高说明过滤人源序列的效果越好,微生物的假阳性就会越低。
图6,人源宿主库三个版本人源序列过滤效果对比。从左到右分别为V1、V2、V3。从总数据量的维度V3相对于V1有0.9%的宿主率提升,而对于过滤人源序列后的剩余数据里面,V3版本相对于V1序列可再过滤50%以上,也即几乎没有人源序列残留。 那么过滤“太干净”人源序列会不会导至微生物漏检呢?也就是原本属于微生物的序列被当作人源序列过滤掉了,导至假阴性。如果人源数据库构建没有问题(没有错误地把微生物基因组加入人源数据库),那完全不用担心。 举一个极端的例子,假设一个微生物的基因组与人类基因组同源的区域有10%(实际上99%以上的微生物物种与人类基因组同源的区域的比例低于1%),如果这个微生物只测到了1条序列,那么这条序列被当作人源序列过滤的概率也就只有10%,测到2条序列2条都被过滤的概率是1%,测到了10条序列10条都被过滤的概率就仅有十亿分之一了。在这种极端的假设下假阴的风险都是极低的。 所以过滤越“干净”人源带来的收益远远大于风险。 微生物基因组数据库构建的挑战 微生物基因组数据库直接决定了mNGS的“检测范围”,而其质量也直接决定了mNGS的结果准确性。
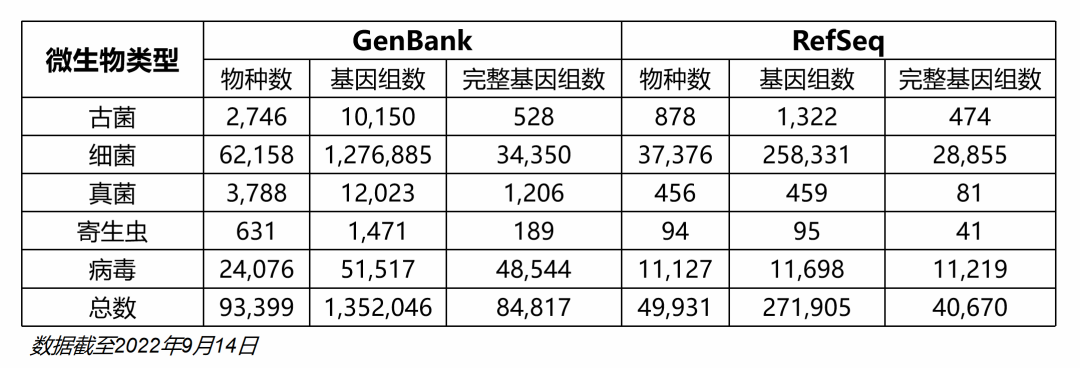
图7. mNGS微生物基因组数据库构建的关键步骤和质控流程。 “基因组数据库从哪里来”也是被经常问到的问题。我的经验就是NCBI的RefSeq、GenBank和Nucleotide(NT库)这三个数据库整合一下就够了,除了新发病原体,或特殊分析目的,否则完全可以不用去折腾其他数据库,因为最后你会发现绝大部分其他数据库的基因组最后都有个“GenBank accession ID”? 但是不是这3个数据库全部都要用?显然不是,一是物种太多,数据量大,二是质量参差不齐。数据库详情具体可以看下面两个表(不是最新数据)。
GenBank是最全的了,也是质量最差的。RefSeq是从GenBank中经过质量评估后筛选,质量稍微好一些,但是真核生物的种类数较少,病毒的亚型也比较少。NT库的最大特点是病毒亚型/株系非常丰富,但是细菌基因组不全,真菌和寄生虫基因组则收录极少。 所以推荐的方案是:以RefSeq为基础,GenBank主要补充真菌和寄生虫,NT库补充病毒亚型和株系,部分病毒细致的分型还需要参考专业的网站,以确保型别的准确性。 FDA-ARGO是美国专为临床诊断用途建立的微生物基因组测序项目。有严格和完善的鉴定和质控流程,测序的基因组质量非常高。项目测序完成的基因组都包含在上述三个数据库中,有覆盖的物种可以优先选择此项目的基因组序列作为代表序列。
对于数据库需要收录多少物种,是不是越多越好?相信关注mNGS的人都有留意到不同的mNGS服务企业宣传的都是2万、3万,甚至4万种以上的。从临床实践的角度看,临床有意义的病原体可能不到3000种,真正有可能检出且报告过的估计最多也就1500种左右。算上一些需要排除干扰的背景菌以及区分同源物种等,5000种也许够用了,1万种就可能几乎覆盖了所有可能性。放再多进去,即使原始列表里面有检出这个物种,那也是一些污染或者算法错误,不是样本真实存在的微生物(仅针对宿主是人的临床样本,非人样本另说)。 当然,前提是你的需要知道这5000或1万种微生物是什么,仅仅依靠经验、文献或者临床书籍是不够的,没有几十万的临床样本作为训练是不具备这个准确选择的能力的。这也是mNGS企业早期追求物种数多的原因之一,因为想知道临床样本里面还有多少未被认识的东西。 微生物基因组数据库构建的真正难点在于质量控制,也就是数据清洗。 微生物基因组除了前面提到的会有人源序列的污染,更严重的还有非人源序列的污染,比如动植物基因组、微生物基因组互相污染,噬菌体污染,人工载体序列污染等等。因为环境中充满了大量各种各样物种的核酸污染,还可能存在试剂污染,交叉污染等等,在微生物测序时原始数据里面就含有这些污染核酸序列,最终被组装进入了微生物的基因组里面并提交到了公共数据库中。
图8. 微生物基因组污染的可能来源。Lu J, Salzberg SL. PLoS Comput Biol. 2018. 同时,做mNGS检测时样本测序数据里面可能也存在类似的污染,因为这些污染通常不会像人源序列一样被提前知道且过滤去除,这就会导至了很多假阳性的产生。真核生物(真菌、寄生虫)基因组这类污染尤其严重。 一个具体的例子就是毛首鞭形线虫(Trichuris trichiura,)至今公开的只有一个基因组序列(GCA_000613005.1),且这个基因组序列还含有大量的大肠埃希菌的序列污染。如果不屏蔽这些污染序列,样本里面有大肠埃希菌检出时可能就会同时带出假阳性的毛首鞭形线虫。 这里特别指出的一个微生物基因组污染噬菌体的例子是:Escherichia phage phiX174(NC_001422.1)常作为illumina测序的平衡文库,被错误地组装进入了大量的微生物基因组中(https://doi.org/10.1186/1944-3277-10-18)。因此过滤Illumina PhiX control是非常有必要的。 一些质粒可以在不同物种间转移,不具备物种特异性。公共数据库的细菌基因组由于组装完整度不同,可能部分物种含有质粒序列,部分物种没有。比如物种A和B都含有同一个质粒,但在数据库里面物种A有这个质粒的序列,物种B没有。如果在检测样本里面单独含有物种B,测序数据里有质粒序列,那这些质粒序列就会被错误分类到A物种去了,最终的检测结果里面就同时检出了物种A(质粒导至的假阳)和物种B(真阳)。此外,质粒还作为基因工程的载体,在实验室中广泛使用,也是一种序列污染源。因此构建数据库的时候质粒序列也应当要考虑去除的。 微生物间的核糖体序列存在高度同源的区域,大部分比对算法对大量高度同源的序列鉴定能力较低,容易发生来源于一个物种的核糖体序列被错误鉴定为另一个物种。这种现象在做RNA检测中尤为明显,如果湿实验环节没有去除微生物的rRNA,那么测序数据里面非人源的数据里可能高达80%以上都是微生物的rRNA的序列,可能会因此产出大量假阳性,特别是真菌的假阳。过滤或屏蔽rRNA序列是值得考虑的。 因此做数据库清洗需要对每一条收录进入数据库的基因组进行污染评估,并且将污染序列过滤或屏蔽。 进行了基因组清洗以后假阳性的检出可以大幅降低(图9)。
图9. NCBI RefSeq数据库清洗前后假阳性检出的对比。左边默认数据库(representative genome),右边为清洗之后的数据库。横坐标为真阳性物种,纵坐标为检出物种,颜色越亮代表检出序列数越高。可以看出左边RefSeq默认数据库有大量假阳性物种检出,而清洗过后的数据库检出的假阳性大幅降低。 但是也可以发现,即使是进行了数据清洗,仍然还是有一定的假阳性结果产生。除了无法完全清洗干净外,还有一些因素是微生物多态性高,且还在持续动态变异、测序错误以及分类鉴定算法等问题。 微生物多样性是分类鉴定的挑战 要做物种分类,首先就要搞明白物种是如何定义的,分类依据是什么?对于动物大家可能都比较清楚:生殖隔离。但是微生物呢?微生物变异速度比较快,特别是RNA病毒,可能几十年的进化过程生物学特征就发生了显著变化。而且即使是同一个物种,它们也是一个庞大的“群体”,各自在不同环境和条件中进化,一直在动态演化着,到什么时候会发生物种级别的分歧?历史上是一个物种,到现在演化了几十上百年,还能算同一个物种吗?
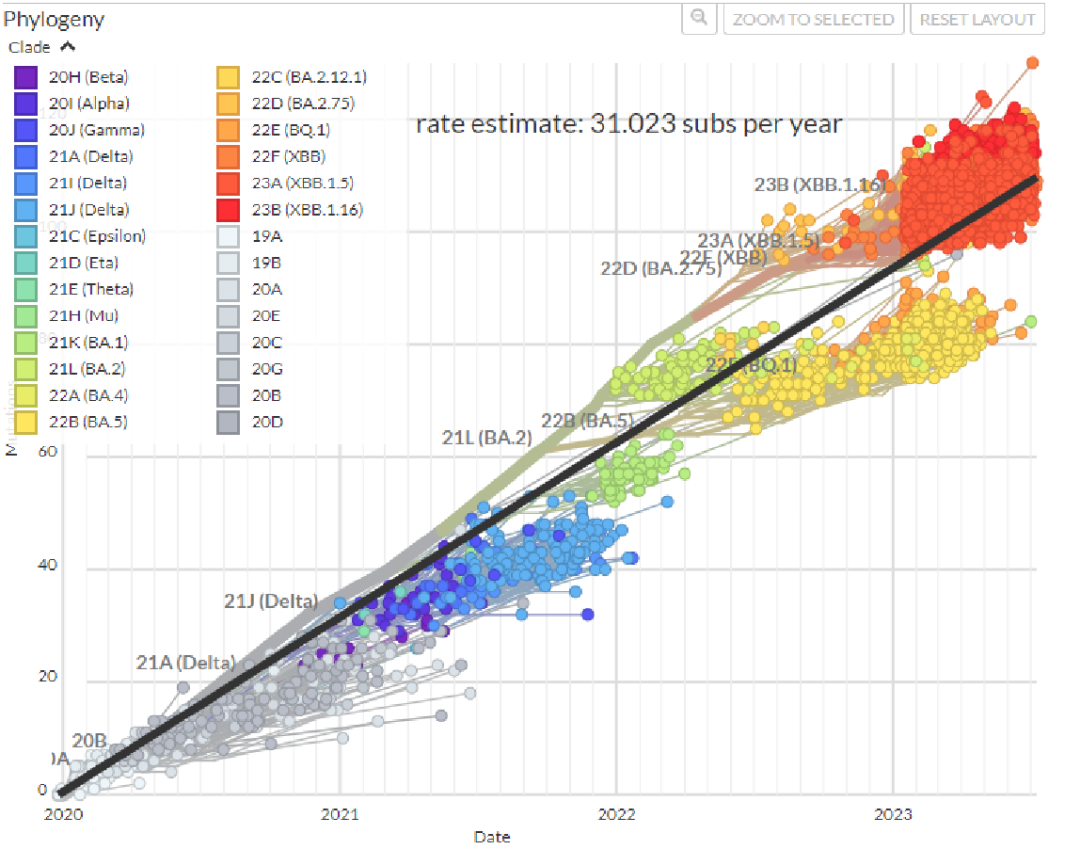
图10. 新冠病毒变异速率约31 subs 每年(NextStrain GISAID数据源)。 传统方法学我们不去讨论,在基因组学上对于细菌来说国际上有个共识:只要基因组平均核酸相似度(Average Nucleotide Identity,ANI)大于95%就可以认为是同一物种。但理想很美好,现实很骨感。虽然大部分细菌都是符合这种定义的,但是由于各种现实的因素,有部分物种却是不合符这种定义的,而且有些还是临床上重要且常检出的物种。 在进化的路上,有些物种在狂奔,有些物种在原地踏步。见下图说明:
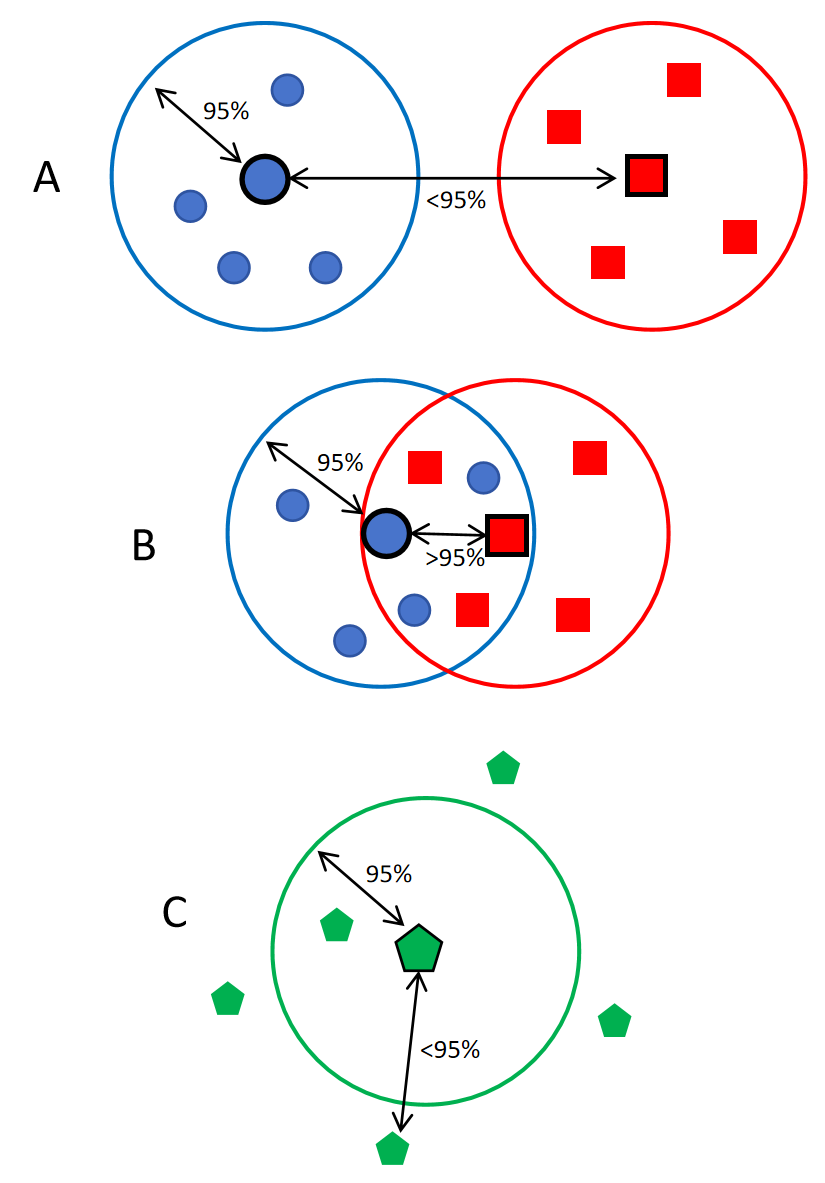
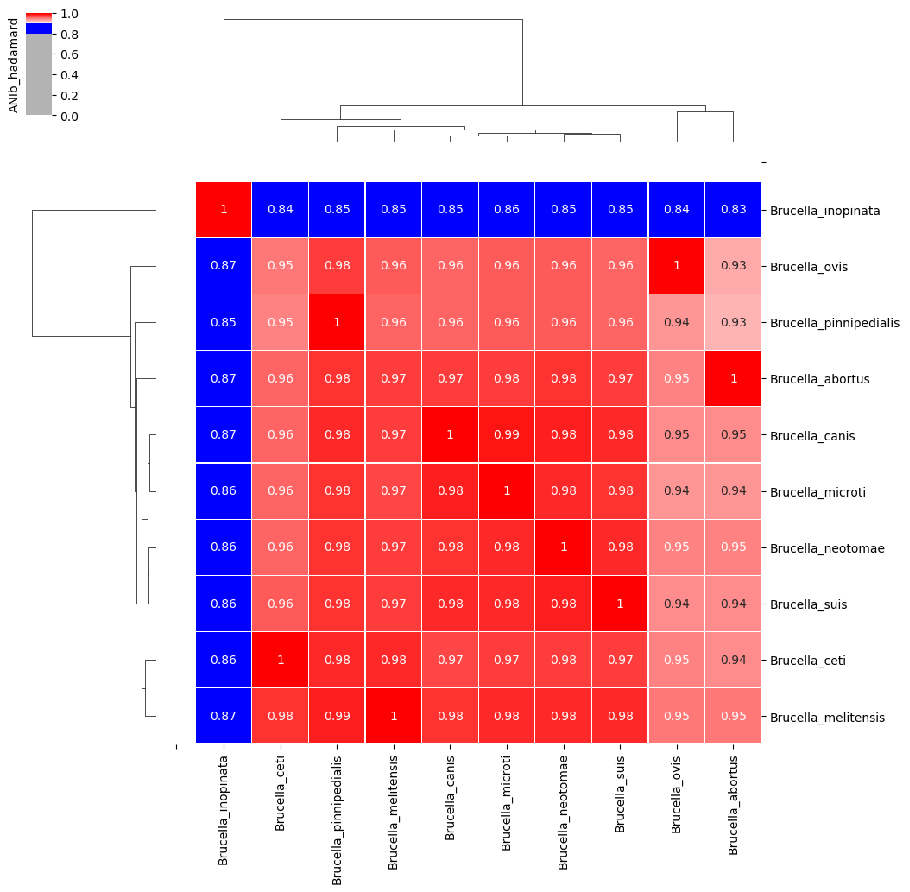
图11. 不同物种间ANI的差异说明。不同颜色代表不同的物种。A. 大多数物种的情况,不同物种间的ANI小于95%,而同一物种内不同株之间的ANI都是大于95%,物种区分度明显。B. 虽然被分类为两个物种,但两个物种间的ANI大于95%,两个物种内不同株之间的ANI也是大于95%,说明两个物种同源性很高,在基因组层面可能无法准确区分。C. 虽然被分类为同一物种,但是不同株之间的ANI小于95%,也就是种内不同株之间差异巨大,甚至可以被当作不同物种。 图11A 代表了大多数物种的情况,种内差异小,种间差异大。 图11C 是少数物种会出现的情况,例如临床常见的嗜麦芽窄食单胞菌(图12)。
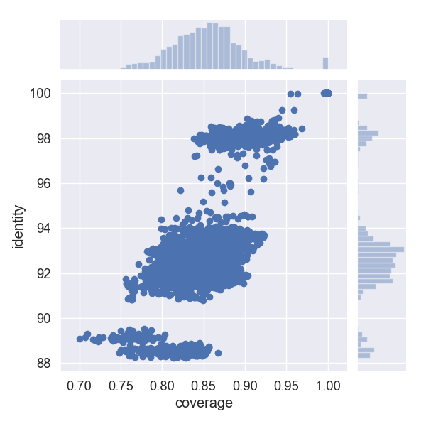
图12. 嗜麦芽窄食单胞菌不同株之间的两两对比。横坐标是两株之间同有区域(Alignment Fraction,AF)的比例,纵坐标是两株之间的ANI,可以看出大部分株两两之间的ANI在94%以下,说明株之间差异巨大,对鉴定会带来一定困难。 图11B这种情况非常典型的例子就是布鲁菌属(图13)、大肠埃希菌和志贺菌属(图14)。
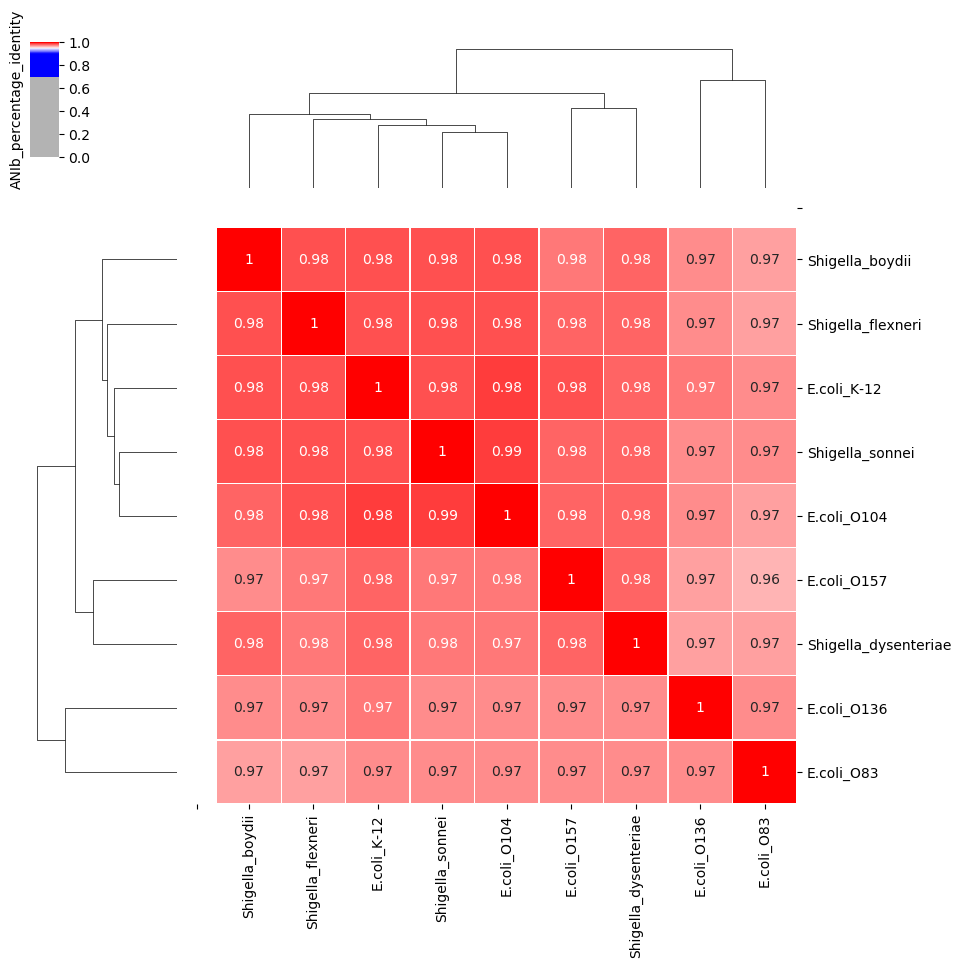
图13. 布鲁菌属不同种之间的ANI可高达98%(个别物种除外),在物种鉴定的时候,布鲁菌基本是不能分类到种水平的,特别是检出序列数比较低的时候。 从基因组ANI的角度,大肠埃希菌和志贺菌属其实是属于同一个物种(图14),很多文献也发表了同样的观点。但是在临床上还是要区分为不同的物种,各有各的依据。
图14. 大肠埃希菌不同血清型与志贺菌属4个种之间的ANI。无论属大肠埃希菌的不同血清型还是不同种的志贺菌,相互间的ANI都高达97%以上。 更头疼的是,从进化树图上看,大肠埃希菌不同血清型的进化距离甚至要大于大肠埃希菌跟志贺菌之间的距离(图15)。
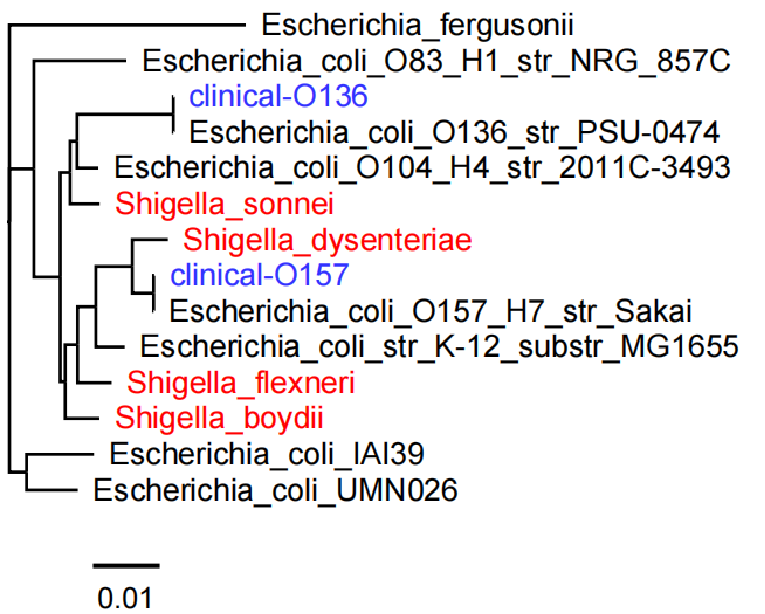
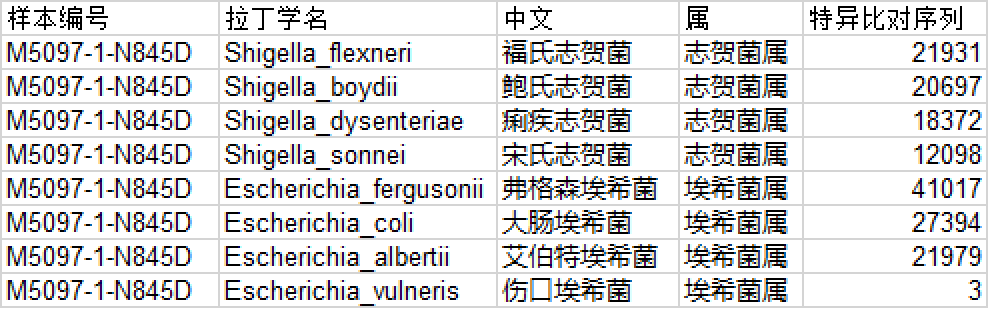
图15. 大肠埃希菌不同血清型与4种志贺菌的进化树。红色为不同的志贺菌,蓝色为临床测到的样本,黑色为公共数据库不同血清型的大肠埃希菌。可以看出4个志贺菌分布在不同的分支上,并且痢疾志贺菌(Shigella dysenteriae)跟大肠埃希菌O157非常接近(ANI的数据也是如此)。 但是,大肠埃希菌和志贺菌鉴定的真正难点不仅在于ANI太高,还在于它们不同株之间特有基因(或区域)丰富。因为ANI计算仅仅是考虑两个基因组共有的区域(同源,能比对上),也就是Alignment Fraction (AF)。大肠埃希菌菌不同血清型之间和志贺菌之间的AF也仅有80%左右(图16)。也就是它们之间的绝对相似度(ANI×AF)仅为80%。任意两两对比会有约20%的序列特有的。
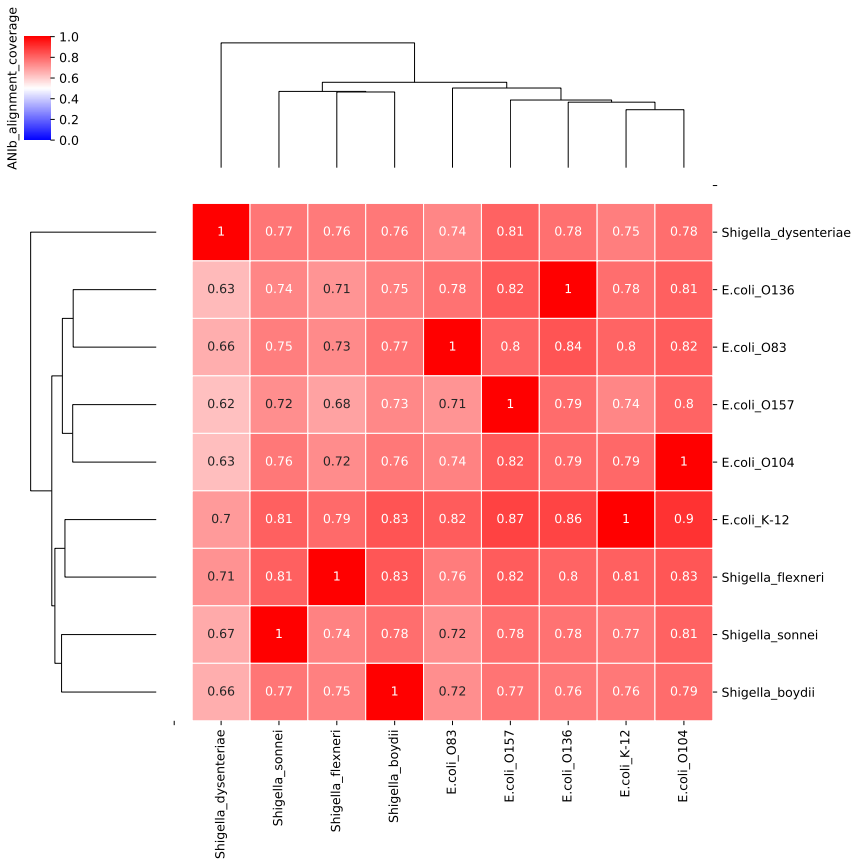
图16. 大肠埃希菌菌不同血清型之间和志贺菌之间的Alignment Fraction (AF). 不同血清型之间AF仅约有80%,与志贺菌之间的AF也差异不大。 这就导至了一个问题就是,一个物种是有很多不同的血清型(或株系)的,而且还在不停的演化,如果数据库里面没有把所有的血清型基因组收录进去,那么会有一部分序列无法比对回属于自己的物种,而这部分没有比对回自己的序列,还有可能因为同源问题错误比对上其他物种去,造成了假阳性。 还是以大肠埃希菌为例,如果数据库里面只收录了一个大肠埃希菌的基因组,比如选择了K12作为代表基因组,而临床样本感染的是O157,因为“O157 vs K12”和“O157 vs 痢疾志贺菌”没有明显的区别,ANI接近,AF也差不多都是80%,所以约80%的序列因为几乎一致而无法区分,剩余20%的序列可能各自比对上了一部分形成特异序列。假设这个样本测到了100条大肠埃希菌的序列,那么最后出来的结果可能是大肠埃希菌5条特异序列,痢疾志贺菌5条特异性序列,无法区分真实的病原体是哪个。 真实样本测出来的结果其实更加复杂。如下面这个例子,临床培养大肠埃希菌阳性。以RefSeq representative genome构建的数据库分析鉴定的结果如下:
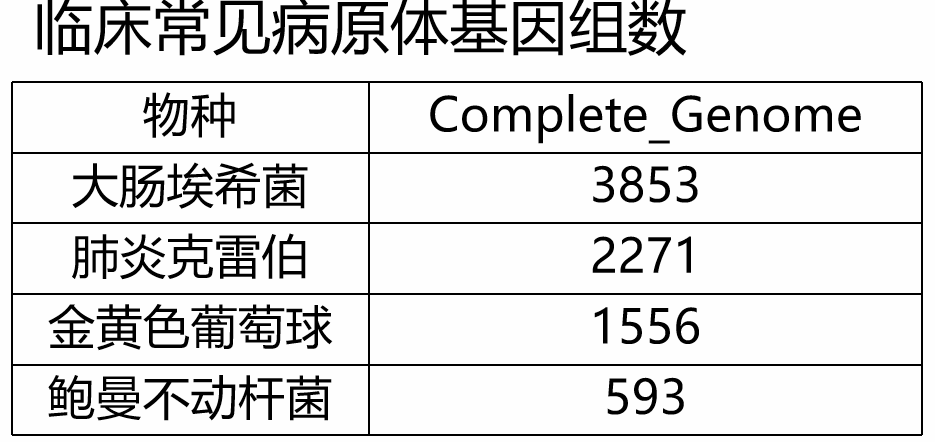
大肠埃希菌出来了,但也同时出来了太多假阳性,大肠埃希菌和志贺菌根本区分不了,且大肠埃希菌的序列也不是最高的。 那这种问题是否可以通过把所有血清型的基因组都放进数据库中去来解决呢?一定程度上可以,但是也有一些难点。比如目前临床上一些重要且常见的病原体,也是研究的热点,大量测序基因组被提交到了公共数据库上,完整的基因组就有好几千条(如下表),全部收录显然不合适的。
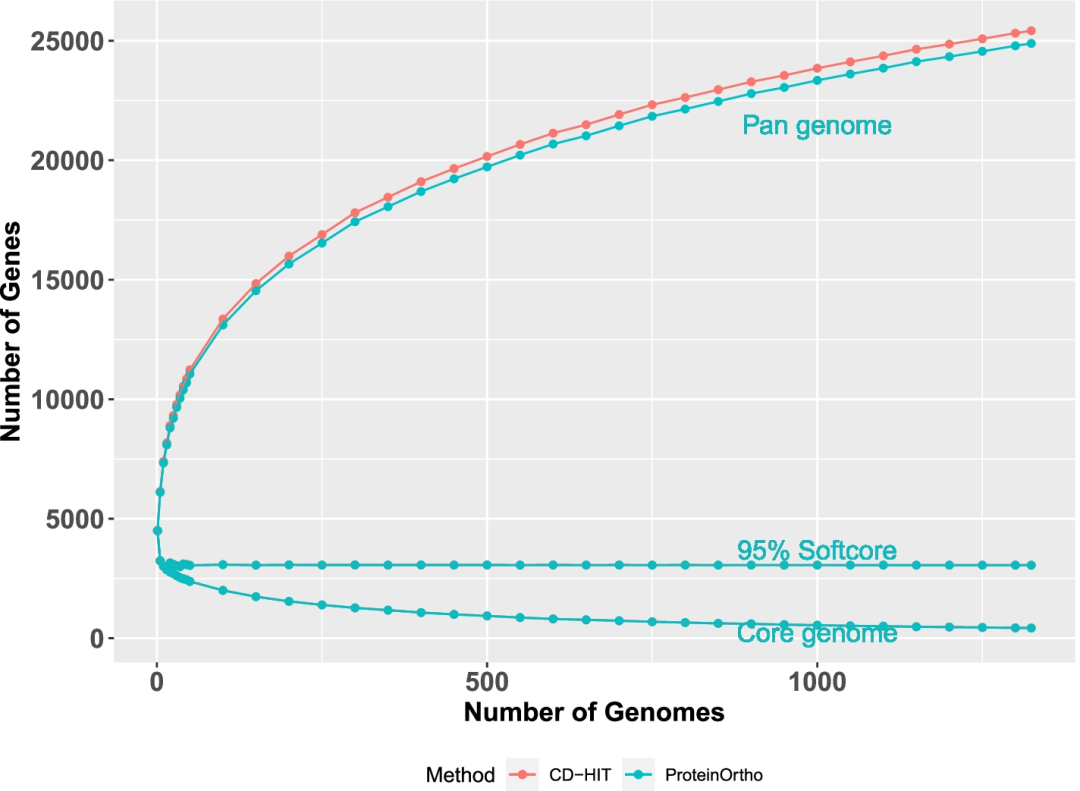
通过pangenome的分析也发现大部分物种的其pangenome仍然会随着基因组数量的增加在无休止的涨,而core gene少得可怜(图17)。
一个物种只选择一个代表基因组不够,选择多了又会造成数据库太大,所以如何取舍又是一个难点。 病原体是存在流行性的,不同时间、不同区域可能流行着不同的株系,新冠就是典型的例子,也被大家所熟知。
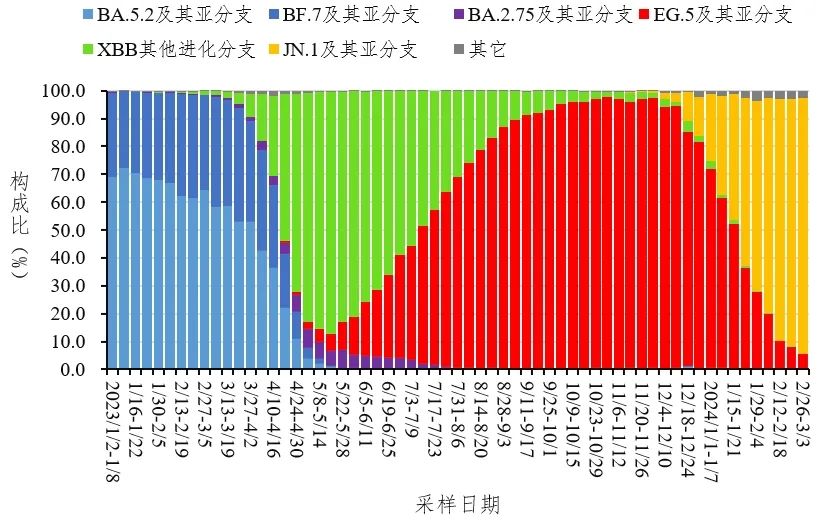
图18. 全国新型冠状病毒感染本土病例变异株变化趋势。中国疾控,2024年2月. 只不过新冠毕竟是一个新的病原体,目前不同株的基因组绝对相似度都还在99%以上,一个原始株基因组就足够准确鉴定了。然而其他病原体都有悠久的历史,其基因组的多样性是巨大的。 如果有积累足够多的临床数据,是可以分析每个病原体流行的株系的,从而可以将流行的株系加入到数据库中。这样做使得数据库中物种基因组有足够的代表性,也可以减少数据库的大小。
图19 大肠埃希菌临床流行血清型前十分布情况。每一个柱子代表一个血清型,不同颜色是不同地区。可以看出大肠埃希菌在临床上没有绝对占优的血清型,多种流行血清型并存。 即便是仅收集临床流行株/血清型,但是大部分物种同时流行的株也不少,株数过多构建泛基因组数据库极其容易积累污染。此外,还有些物种甚至是有错误分类的株,需要仔细评估再选择。
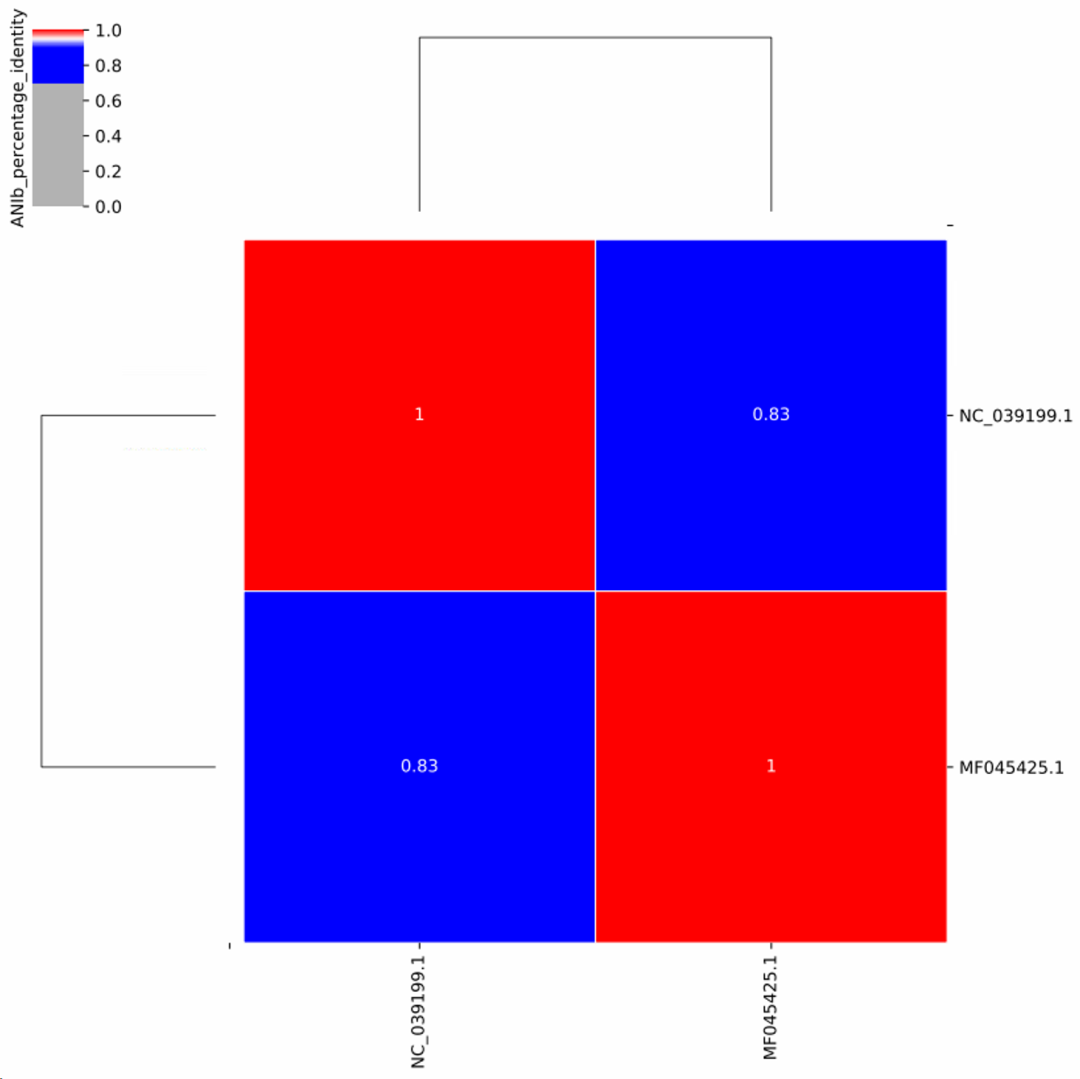
图20. 同一个物种不同株之间的相似度关系。图中有小部分株与其他株差异巨大,不排除是错误分类的株,基于谨慎原则,应当舍弃。 在临床上,RNA病毒是最容易因为流行株的不同导至鉴定出问题的。例如临床上经常出现的人类偏肺病毒(Human metapneumovirus),RefSeq的代表基因组是NC_039199.1,但实际临床流行的几乎都是以MF045425.1为代表的株系,而NC_039199.1与MF045425.1两者的ANI仅有83%(图21)!如果数据库里面只用了RefSeq的代表基因组NC_039199.1,ANI差异如此之大,比对软件是比对不上的,要么直接漏检,要么检出很低序列。其他还有一些RNA病毒也有类似的情况,如鼻病毒等。
图21. 人类偏肺病毒(Human metapneumovirus)RefSeq的代表基因组NC_039199.1与临床流行株MF045425.1的ANI差异,两者ANI仅有83%。 除了要进行分类评估,还有前面提到的,很多基因组是有片段污染的,即便是组装度为“完整”的基因组也可能有污染,泛基因组最容易积累这些污染。例如鹦鹉热衣原体在GenBank上有几十株基因组,其中有几株是有人源序列污染的,且集中在某个实验室提交的数据,不过其组装完整度一般,需要注意将这类数据排除。 即便做了非常多的质控,最后构建出来的数据库在分析临床数据时还是有可能会发现一些新的污染问题,甚至可能有一些目前尚未测序的动植物或微生物的污染,这些未知的序列也可能错误比对到数据中的物种去,造成假阳性。因此需要通过大量临床数据去积累和过滤这类污染。 进行了这么一系列的分类质控、去污染、流行株筛选、泛基因组等等的操作,到底有没有用?以前面大肠埃希菌培养阳性的样本为例子对比一下: RefSeq代表基因组构建的数据库结果:
自建数据库的结果:
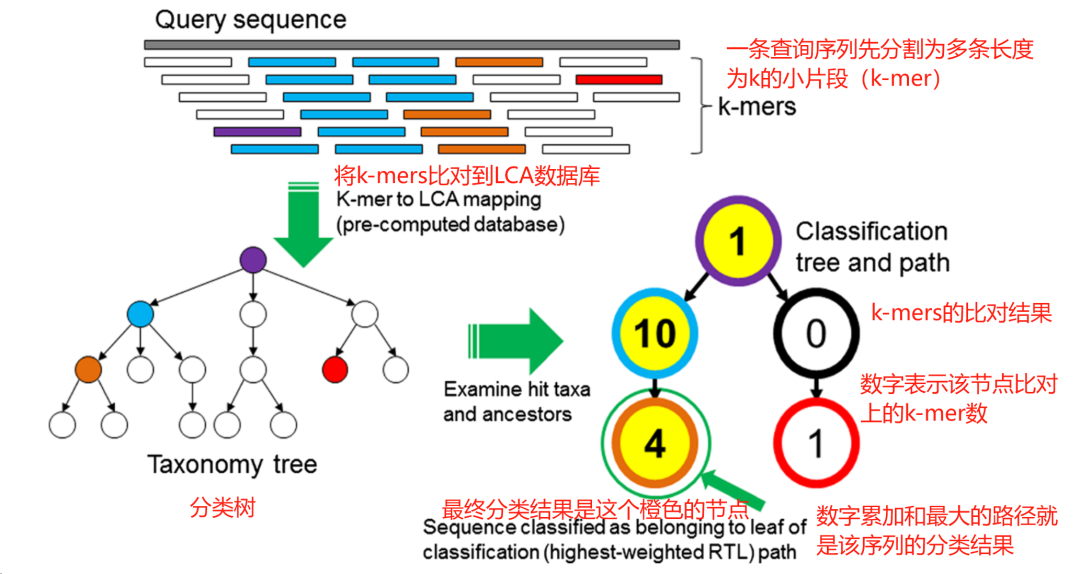
自建数据库的信噪比远远大于RefSeq代表基因组数据库,这就是差距。 但是可以看出这个结果仍然不完美,还是有其他假阳性的物种,尽管是很低比例的。这里的原因除了数据库没有办法做到完美以外,还有就是分类算法的问题。 分类鉴定算法如何影响mNGS的结果 mNGS的物种分类鉴定具体来说包括两个步骤:比对和分类。 序列比对 在序列比对环节,目前主流的有两种算法:基于k-mer的比对算法和基于动态规划的比对算法。基于k-mer的算法通常是快速且高效的,由于它们不需要在序列间进行逐个碱基的比较,即没有执行传统意义上的alignment过程,因此这类方法也常被称作alignment-free算法。而基于动态规划的算法则能够容忍序列中的一定错误或变异,动态规划算法用来执行局部比对(local alignment)或全局比对(global alignment),因此这类方法被称为alignment-based算法。 基于k-mer的算法,是以精确匹配为前提的,只有当两个k-mer片段(即查询序列中的片段和参考序列中的片段)完全相同,才被视为匹配成功。在比对过程中,一条序列会被拆分成多个长度为k的子片段(k-mer),然后通过评估这些子片段与参考序列中的k-mer匹配的比例来判断整个序列是否与参考序列相符。科研领域广泛使用的Kraken就是使用这种算法的代表性软件。基于k-mer的方法主要优势在于其分析速度快,适合处理大规模序列数据。但这种方法主要缺点就是它不具备容错能力,不适用于变异较大的微生物,在实际的使用上性能表现较差。 基于动态规划的可容错比对算法,其核心步骤通常涉及初始定位和逐步精细化的两个阶段。首先,算法会将查询序列分割成若干较短的子串,称之为'seeds',即种子,然后通过这些'seeds'在参考序列中快速定位潜在的匹配区域。定位完成后,算法使用构建得分矩阵的方式进行动态规划,详细评估整个查询序列与参考序列间的匹配情况,以找到最佳比对路径。动态规划过程包括对匹配、错配以及插入和缺失(即gap)的情况进行积分和罚分,从而得到总体得分最高的比对结果。不同的软件可能采用不同的种子选择、索引构建及打分过程来优化搜索效率和比对质量。BLAST、BWA、和Bowtie2等是这类算法的代表软件。容错能力是这类算法的显著优势,使其能够灵活处理序列中的变异。此外还可以输出序列比对的详细信息,这些信息对后续的物种分类极为重要。目前几乎所有mNGS厂家都使用这类算法。 物种分类 要做物种分类,首先要有物种分类数据库,即物种的命名以及在分类层级上的关系(界、门、纲、目、科、属、种等),某些病原体可能需要分类到种以下的水平,特别是病毒,通常需要分类到种以下的型或亚型。常用的分类数据库是NCBI taxonomy,但这个数据库不是国际权威的数据库,有些物种的命名或分类可能需要参考更加权威的分类数据库,如病毒的ICTV,细菌的LPSN。 序列比对之后一般采用Lowest Common Ancestor (LCA,最近公共祖先)算法进行分类。 在Kraken软件中,序列比对和LCA分类是一起完成的,参考数据库是预先构建好的包含了LCA信息的数据库。一条序列的最终分类结果是根据其k-mer的分类结果确定的。
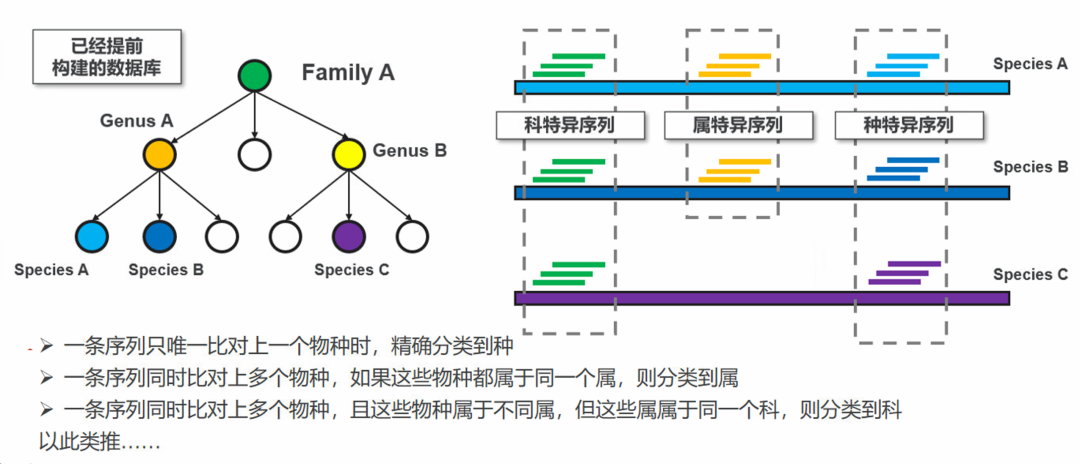
图22. Kraken分类算法。一条序列会先拆分为多个k-mer,每个k-mer都被比对到数据库中包含该k-mer的基因组的最近共同祖先(LCA)。在分类树中,每个节点的权重等于k-mer数量。每条从根到叶子(RTL)路径累加该路径下节点的得分,得分最高的路径就是这条序列的最终分类,如果有多条得分一样高的路径,则这些路径的LCA为最终分类结果。 使用BWA、Bowtie2这些软件则需要自己开发分类算法,因为这些软件只起比对作用。难点也是在这里。 首先就是如何定义一条序列对上了参考基因组?理想的情况下两条序列100%一致当然可以算比对上了,但是微生物基因组是有多态性的,用BWA这些软件也是因为其具备容错比对的能力,那如果两条序列不是100%一致的时候,一致率是多少才算比对上?目前这个没有统一的标准,但这个阈值是可以显著影响最终的分类结果的。 以细菌为例,前面提到过同一个物种的共识是ANI大于95%,属内不同物种的ANI在80%左右(不同属差异大),在临床上通常要分类到种,甚至种以下的水平。所以对于细菌来说,比对上的阈值设置为ANI大于95%也许是一个合理的选择。但是对于病毒来说,特别是一些多态性高的RNA病毒,则可以设置更低的阈值。不同的分类层级也可以设置不同的阈值,但是受限于比对软件的容错率能力,这个阈值下限也不可能太低。要追求极致的性能,需要根据每个物种基因组的多态性水平进行个性化设置。 定义好一条序列“比对上”之后,接下来就是对这条序列进行LCA分类了。与Kraken不同的是,基于BWA这类软件的比对结果做分类是直接对这条完整的序列做LCA。即:如果一条只比对上一个物种,那么这条序列可以直接精确分类到这个物种。但一条序列也可能同时比对上多个物种,这个时候这条序列就分类到了这些同时比对上的物种的最近公共祖先,比如属、科等等。
图23. LCA分类算法说明。 左边:物种A、B、C是不同的三个物种,在同一个科,其中A和B还在同一个属。右边:绿的序列同时比对上了物种A、B、C,他们的最近公共祖先是科A,则这些序列被分类到了科水平。黄色的序列同时比对上了物种A和B,物种A和B的最近公共祖先是属A,则这些序列分类到了属水平。青色、蓝色、紫色的序列分别只比对上了物种A、B、C,所以他们分类到了种水平。 比对算法和分类算法如何影响最终的分类结果? alignment-besed的比对算法其参数(种子长度、匹配、错配、gap的得分和罚分数等)是可以直接影响一条序列的比对结果的。一条序列在多物种比对的时候如何判断阈值也是可以显著影响分类结果。特别是对于高度同源的物种,在数据库层面无法解决的情况下,只能依靠分类算法进行优化。 首先来看一些例子。 例子1:假设数据库中有两个近缘物种A和B,基因组某个区域其序列如下,这两条序列仅有一个碱基差异:
将来源于样本的测序数据与这个数据库比对,一条查询序列与物种B完全相同,理所当然,这条序列应该被鉴定为物种B。但这种判断一定没有问题吗?
接下来的例子就是这种的判断方法在实际应用场景里面遇到的问题了。 例子2:如果临床样本里面真实存在的病原体是物种A,但很不幸,由于测序错误或者变异,物种A测到的序列原来是G的碱基变成了A。
但是参考数据库里面物种A那个位点的碱基还是G,而临床测序的序列此位点的碱基由G变成A后与物种B的序列完全一样了,那么按照例子1的判断逻辑,此时这条序列就会被鉴定为了物种B。
所以例子1里面的判断逻辑上遇到同源性高的物种的时候就容易出现假阳性。当然,因为微生物多态性或者测序错误带来的这类错误鉴定的序列比例还是比较低的,所以真实临床样本如果只含有物种A,那么最终的检测结果里面可能就是大部分序列还是物种A的,少部分序列被错误鉴定为物种B,也就是A和B同时检出,其检出的序列比例取决于数据库构建质量、基因组本身的多态性和测序错误率等。理想的情况下可能是个位数的以下的百分比,这种情况如果有经验还可以识别出来,但夸张的时候甚至可以达约1:1,这种情况就基本无法判断真实的到底是A还是B,还是两者都有了。 例子3:如果这条查询序列刚好与物种A和B都相差一个碱基,那么这个时候这条序列该鉴定为什么?
如果比对阈值是允许一个碱基错误的话,那么这条序列就是两个物种都算比对上了,因此这条序列就鉴定为物种A和B的LCA。如果阈值是不允许错配的话,那这条序列只能当作不可分类序列。 例子4:如果数据库仅包含物种A,且一条查询序列与物种A的基因组仅存在一个碱基的差异,并且我们接受一个碱基的错配,那么这条序列就会被归类为物种A。
然后在真实应用中又遇到问题了。此时更新了数据库,新加了一个物种C,发现这条查询序列可以与C完全相同,原来分类为A的序列,此时就应该分类为C,或者A和C的LCA。
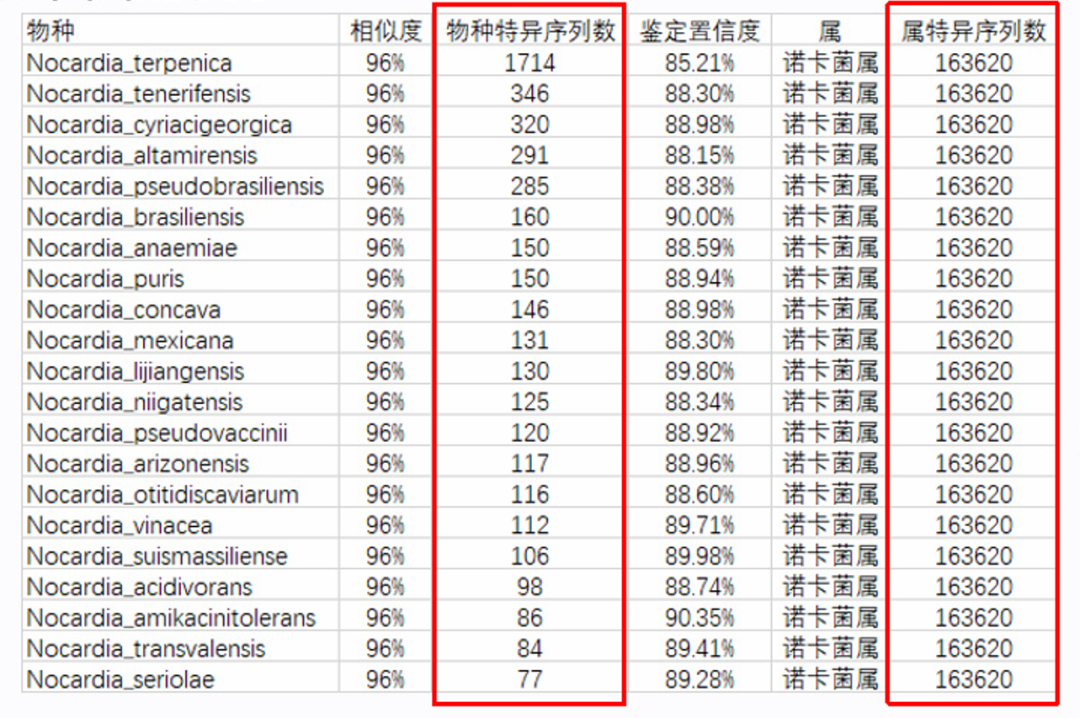
这种情况就是数据库不全可能会经常遇到的问题,如果样本里面有的病原体,在数据库中没有参考基因组,那么就会错误分类到近缘物种,但因为近缘物种可能有多个,所以就可能检出多个假阳性物种。在临床检测中,诺卡菌、分枝杆菌、部分真菌可能比较容易出现这种情况。因为这些菌属可能还有比较多的一些物种还没有被发现和测参考基因组。比如下面这个疑似检出未知新种诺卡菌的例子:分类到属的序列很高,但是分析到种的序列很低,且同时检出多个种,每个种的序列差异不大,ANI也不高。
怎么去处理这些难题,以得到更加准确的结果呢?一些可能的思路是调整比对参数和调整分类逻辑。 调整比对参数 调整比对参数需要对比对算法和所用软件参数有深刻认识,知道其底层原理是如何影响最终结果的。 这里以BWA MEM为例,BWA MEM算法的核心是使用BTW构建索引,比对过程包括通过seed进行潜在位置匹配、seed扩展(Local Alignment)等,这些步骤涉及的一些参数是关键的,如:
一些结果输出的参数也是对后续分类鉴定有影响的,如:
假设有这么两条序列,来看看不同的参数如何影响最终的比对结果:
参考序列长51bp,查询序列长50bp。查询序列与参考序列有两个不同,一个是第18位有个替换(A>C),一个是尾部有个单碱基缺失(C>)。使用BWA MEM默认参数比对结果如下:
即:
比对的结果是直接将查询序列尾部的2个碱基给切除了(软切),因为软切罚分仅5分,而间隙开放罚分6,错配罚分4+4=8,因此得分最高的是软切策略。这种情况查询序列比对长度是48,同时有一个错配,其ANI是47/48=97.9%,如果以95%作为比对阈值的话,这种情况还算是比对上了。 如果我们调整参数,将错配的罚分降低至1分(使用-B 1参数),则会出现以下比对结果:
即:
尾部两个碱基直接当错配了,因为错配罚分是2,而软切罚分是5,间隙开放罚分6,得分最高的是错配策略。这种比对结果总共有3个错配,ANI是47/50=94%,过不了95%的阈值,所以这条序列不能算比对上。 如果把间隙开放罚分调小,设置为只罚1分(-O 1),就会出现下面的情况:
即:
尾部的比对出来了gap,因为参考序列是两个连续的C,实际上这种比对结果跟最初假设的结果是等同的。间隙开放罚分1,错配罚分是2,软切罚分是5,得分最高的是间隙开放策略。这种比对结果是1个错配和一个单碱基的gap,ANI是48/50=96%,可以过95%的阈值,比对上了。 如果调整种子最小长度,设置为31(-k 31),这是kraken默认的k-mer长度,就会出现下面的情况:
输出结果第二列flag值为4,即无比对结果,在比对软件层面这条序列直接无法比对上参考序列。这是因为两条序列在第18位有个错配,当最小种子长度是31的时候,查询序列生成的所有长度为31的种子里都会至少含有1个错配,而在比对过程中种子是精确匹配的,因此在种子匹配环节这条序列就比对不上了。 所以,如果用Kraken软件,且默认k-mer长度是31,那么这条序列就会无比对结果。 可以看出,同样的两条序列,即使同一个软件,不同的比对参数设置就得到了不同的比对结果,甚至直接决定着一条序列是否能比对上。所以在实际应用场景中需要不断调整比对参数进行测试,根据最理想的结果,找出最优的比对参数。 这种比对参数的不同在实际应用中会带来多大的影响呢?以呼吸道常见的病原体流感嗜血杆菌为例。 使用BWA MEM的默认参数(以ANI大于95%为物种分类阈值):比对上序列数是31,415。
优化比对参数后:比对上的序列数是34,775,提升了约10%。
仅仅是比对参数的调整就带来10%的信号值提升,但更重要的是,这10%的序列在默认参数的时候比对不上流感嗜血杆菌,但它们会可能错误比对上其它物种去,导至出现假阳性的结果。 比对参数之后,分类算法对分类结果的影响更大,特别是高度同源的物种。 序列是唯一比对时,可以直接分类到比对上的物种,序列多比对时(同源物种经常出现的问题)就需要用LCA进行分类。但是对于一些同源性高的物种,如果使用统一固定的ANI作为阈值,那么就可能出现要么分类到种的序列特别低,导至灵敏度低;要么出来很多同源的假阳性物种,导至特异性低。针对这些问题,可以个性化调整ANI阈值,还可以采用序列与不同物种的ANI的差异进行判断,甚至采用更为复杂的机器学习方法,如贝叶斯估计。 还是以流感嗜血杆菌为例,由于它与属内的埃及嗜血杆菌、溶血嗜血杆菌、副流感嗜血杆菌等在基因组上具有较高的同源性,而这些同源物种通常是常见的呼吸道定植菌。因此,如果分类不准确,就可能导至错误的将这些定植菌识别为流感嗜血杆菌,从而产生假阳性结果。 如果直接使用比对软件出来的默认结果且不做LCA分类,结果如下:
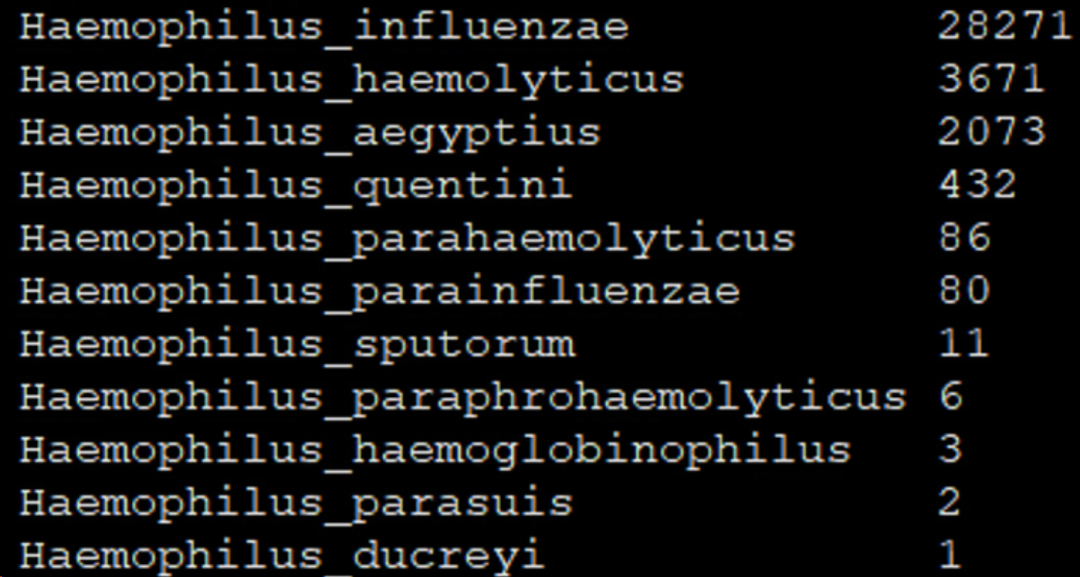
除了流感嗜血杆菌这个真阳性以外,出现了多个属内的假阳性,序列数还不低。真阳性与最高序列数的假阳性的信噪比是:28271/3671≈7.70 做了LCA分类之后:
真阳性的序列数有所下降,但假阳性的序列数降低幅度更大,假阳性物种的数量也减少了。真阳性与最高序列数的假阳性的信噪比是:20534/160≈128.33,比原来提高约17倍! 做了LCA分类+分类算法优化后:
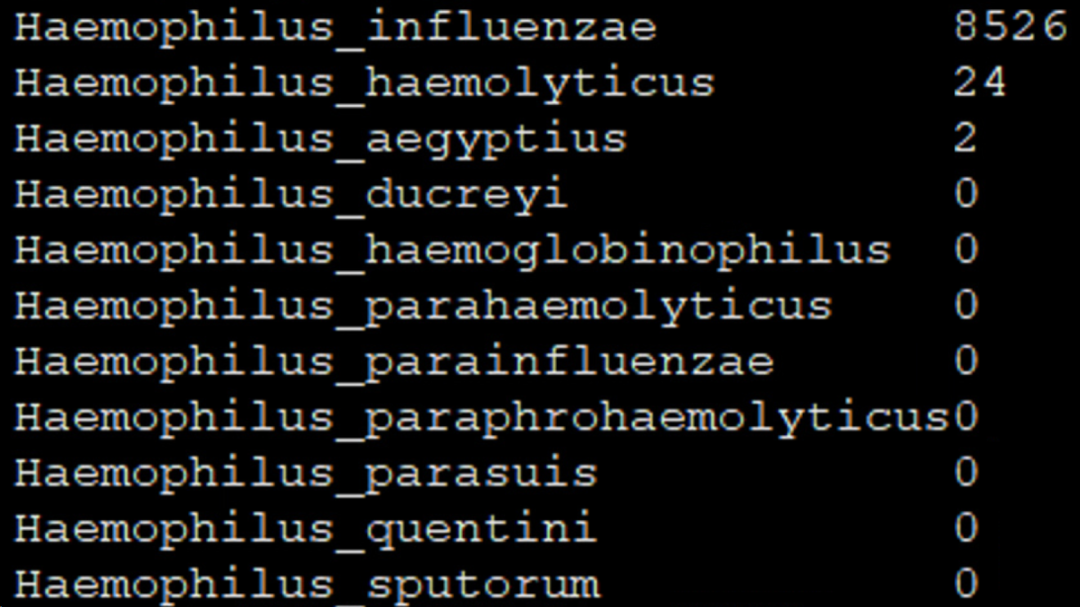
真阳性的序列数还是有所下降,假阳性的序列数也以更大的幅度下降,假阳性物种也降低到了2个,有个假阳序列数也只有2条。真阳性与最高序列数的假阳性的信噪比是:8526/24=335.25,比仅采用LCA算法再提高约3倍。 需要说明的是,不同的物种可能有不同的模式,另外也可能需要在信号值和信噪比之间做一些平衡,在物种检出序列数低的时候对灵敏度有一定的影响。 其他一些属,如链球菌属、葡萄球菌属等也存在属内不同的物种有些是定植,有些是致病菌,且同源性比较高的情况。特别是定植菌通常丰度还很高,分类算法对同源性物种区分不好时,极容易带出假阳性的致病菌。部分临床上重要的同源性高的物种如下,红色字体的是临床比较常检出或容易鉴定错误的。
从上面的例子中也可以看出,尽管基因组数据库、比对算法、分类算法都做了深度优化,但还是有一些假阳性结果,还有没有什么解决方案了呢? 一种可能的策略是对每个检出的物种引入“鉴定置信度”打分。 mNGS理论上是一种无偏倚测序方法,对于真阳性的病原体,其测到的序列落在基因组上任意位置的概率符合泊松分布,在不考虑GC含量等差异的影响下,也就是基因组任意相同长度的区间检出的序列数是相近的。直观上的感觉就是检出来的序列在基因组上图画出来的分布图是“均匀的”。例如长这样:
图24. 真阳性物种检出序列在基因组上的分布。基因组分为50个区域,每个区域比对上的序列数无显著差异。 而假阳性的物种是有一定特征的,要么是片段污染,要么同源鉴定错误等。污染的片段、同源的区域都是一些比较小的片段,所以假阳性的序列基本都是出现在这些位置上,是不符合泊松分布的。那么画出来的基因组分布图是很“不均匀的”,例如长这样:
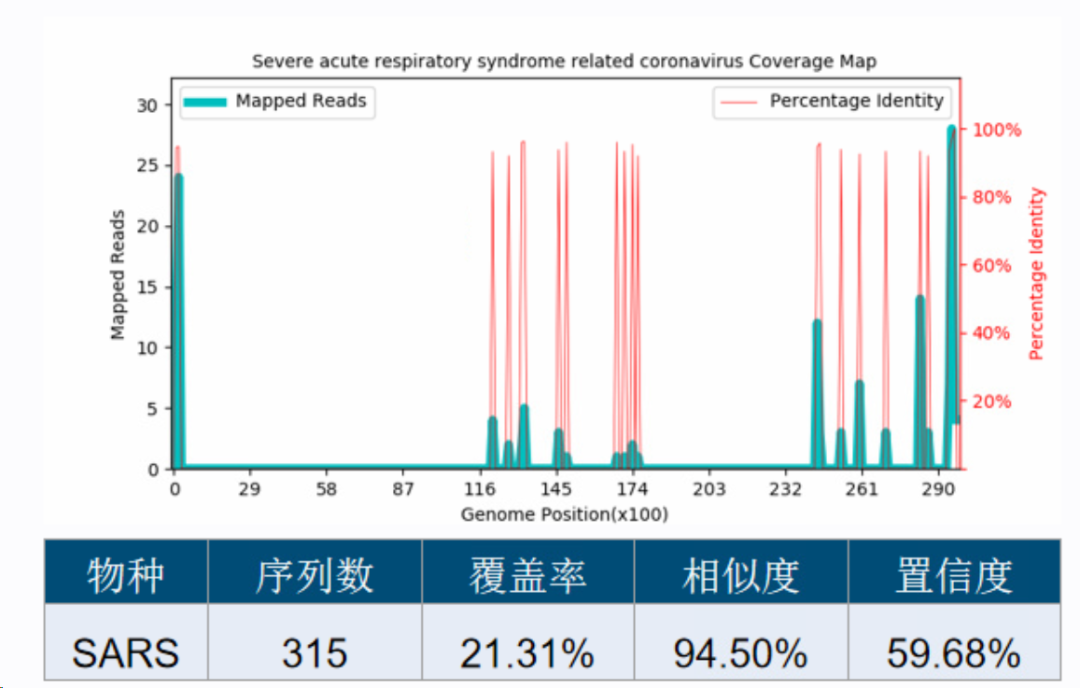
图25. 假阳性物种检出序列在基因组上的分布。基因组分为50个区域,每个区域比对上的序列数差异显著。 这种分布的随机性可以用“离散度”来衡量,但是仅仅这个离散度还是不够的,特别是做RNA检测时,本身基因的表达是有差异的,在测到的序列在基因组上不同的基因区域出现的概率不相等,所以还需要引入其它指标,例如检出序列数、ANI、其它物种的检出、同源物种的先验信息、测序深度、宿主率等等指标,进行不同权重的打分建模,训练出性能可用的模型来判断。这也是一个大工程,没有统一标准,不同人有不同思路和经验,不展开讨论。 以新冠病毒为例,如果数据库没有新冠病毒的序列(比如刚发现新冠时),就会检出来SARS-CoV的假阳,如下图,检出序列数较少,序列在基因组上分布不均匀,ANI仅有94.5%,最终算出来的置信度只有59.68%(90%以上为可信)。
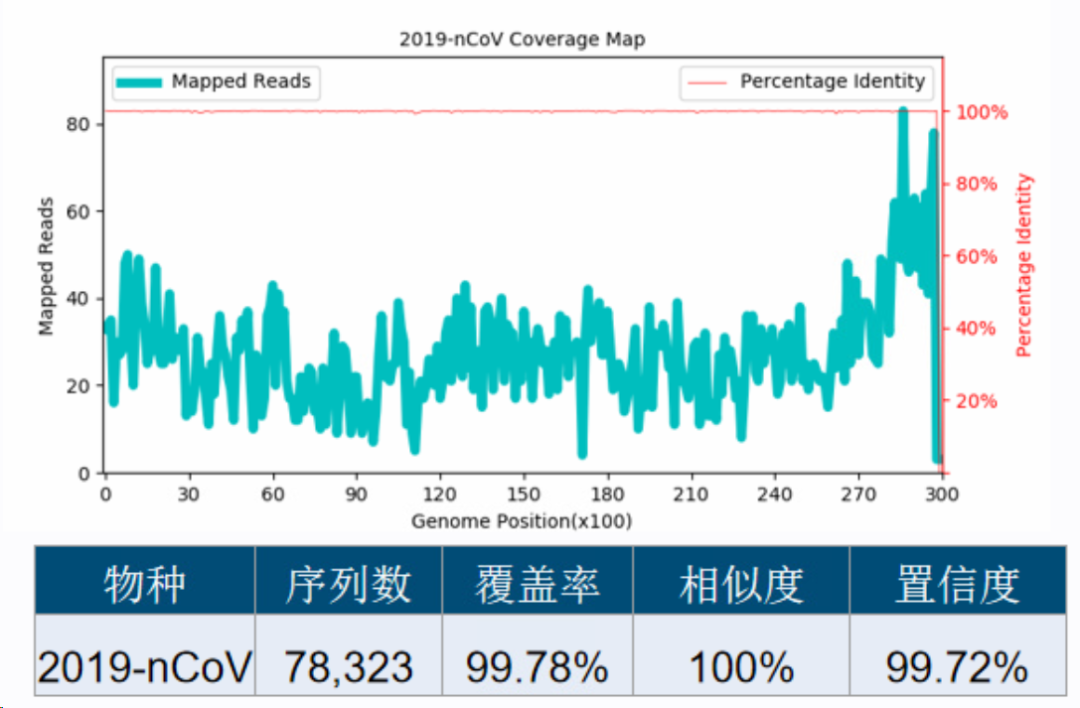
图26. 新冠病毒导至的SARS-CoV假阳性检出。只有部分同源性高的区域有序列检出,每个区域检出的序列数差异显著,最终算出的置信度也低。 数据库有了新冠病毒以后,检出来就是新冠了,序列数高达78,323,序列在基因组上的分布比较平均,ANI为100%,最终算出来的置信度是99.72%。
图27 新冠病毒真阳性检出。每个区域检出的序列数无显著差异,最终算出的置信度也高。 所以鉴定置信度不仅可以作为假阳性物种的判定指标,还可以作为发现新发病原体的线索! 数据库优化+比对参数优化+分类算法优化+鉴定置信度基本上可以解决大部分的生信假阳性问题了。但是对于mNGS而言,生信假阳性仅仅只是假阳性的一部分,还有很多在湿实验环节引入的假阳性,也就是背景菌或污染菌。其来源可能是采样污染、实验室环境污染、试剂工程菌、实验耗材污染,甚至是测序环节的barcode污染、index hopping等等。这些假阳性物种其核酸是真实存在的,也就是从生信的角度看,这些物种的检出是真实的,但从样本的角度来看它们还是属于假阳性的物种。 这个时候只能在下一个环节处理了——报告解读。也就是区分检出的微生物是属于mNGS流程种引入的实验噪音,还是真实来源于样本的微生物,来源于样本的微生物又还可以区分为定植菌(人体微生态)和致病菌,甚至还可能检出一些意义未明的菌。
图28. mNGS报告原则。 实际上,报告解读可以分为‘技术解读’和‘临床解读’两个层面。但由于mNGS方法学的局限性和对mNGS认知的不足,这两个层面的解读往往交织在一起。 何为“技术解读”?实际上技术解读还是属于生信的范畴,任务就是要排除样本检测结果里面的实验噪音,也就是排除生信假阳、污染菌/背景菌等。这个就是检验方所需要做的事情,保证报告出去的微生物都是真实来源于样本的,同时也不能漏。理论上就是对样本负责,样本有什么就报什么,不错报、不漏报。但是在经验上,有些已经非常明确的定植/微生态、严格致病病原体,还是会做一些提示或者分模块呈现。 “临床解读”就是假设在检出的微生物都是真实的前提下,根据检出的结果,结合患者的临床信息,判断责任病原体以及确定治疗方案。特别是对一些“混合感染”、条件致病病原体等,非常依赖医生经验和患者的临床信息,由医生来判断是哪些是责任病原体,哪些需要用药等等。 上面说的是理想的情况下,检验做检验的,临床做临床的,各自职责明确。但在现实中,首先“技术解读”就没有办法完全做到排除“实验噪音”,质量控制没有做好的时候,“实验噪音”是五花八门的,甚至有一些是在样本采集环节的污染,仅依靠技术或者经验是无法解决的。 特别是遇到了一些既是临床重要病原体,又无法排除“实验噪音”的时候,技术解读就会想问问临床:同志,有这么个病原体,从患者的临床信息上看,你们觉得符不符合?也就是需要结合临床信息来确定检验的结果。 相反,有些时候临床的医生对于一些检出的病原体,如果在自己意料之外,或者可能跟患者临床症状不符,反过来也会找技术人员:同志,这个东西到底真不真,再帮忙看看?结合了技术解读的信息后,就会增加临床医生的判断信心。 其实这些就是朴素贝叶斯的思想,也是智能报告系统的方向之一。 生信在这一环节的挑战就是要解决“技术解读”所面临的难题,但是“技术解读”所面临的难题又涉及mNGS整个流程的问题,特别是mNGS全流程的质量控制问题,比如污染严重、实验不稳定甚至失败等等,严重影响了检出结果稳定性,生信也是无力回天的。 总结 mNGS生信面临的挑战主要包括“全面、准确、快速、经济可负担”等要素,并且需要全部兼顾。通过对基因组数据库全面地收集(宿主和微生物)来保证“全面”。通过严格的基因组质量控制、物种比对和分类算法优化、引入鉴定置信度等来保证“准确”。通过对基因组特征深入分析和临床大数据训练流行株等方式来保证基因组全面的同时还能做到“精简”,减少数据冗余,缩短了分析时间和降低硬件资源需求,实现了“快速和经济可负担”。 但mNGS生信仍然还面临很多挑战,还有大量未经测序的物种,数据库的全面性、基因组的质量控制无法做到尽善尽美,微生物基因组多态性及动态变异、物种高度同源、测序错误等带来的假阳性也无法完全仅通过算法来解决。这里所罗列的挑战也仅仅是众多的挑战中的一小部分。 此外,mNGS是涉及多环节的整体技术,整体技术层面的质量控制面临的挑战远大于生信环节的挑战,生信不可能孤立起来解决问题,还需要联合湿实验、临床等多方,从整体流程来解决mNGS所面临的挑战。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号