任何疑问、批评、指导,请毫不犹豫地私信作者!
研究方法
研究结果
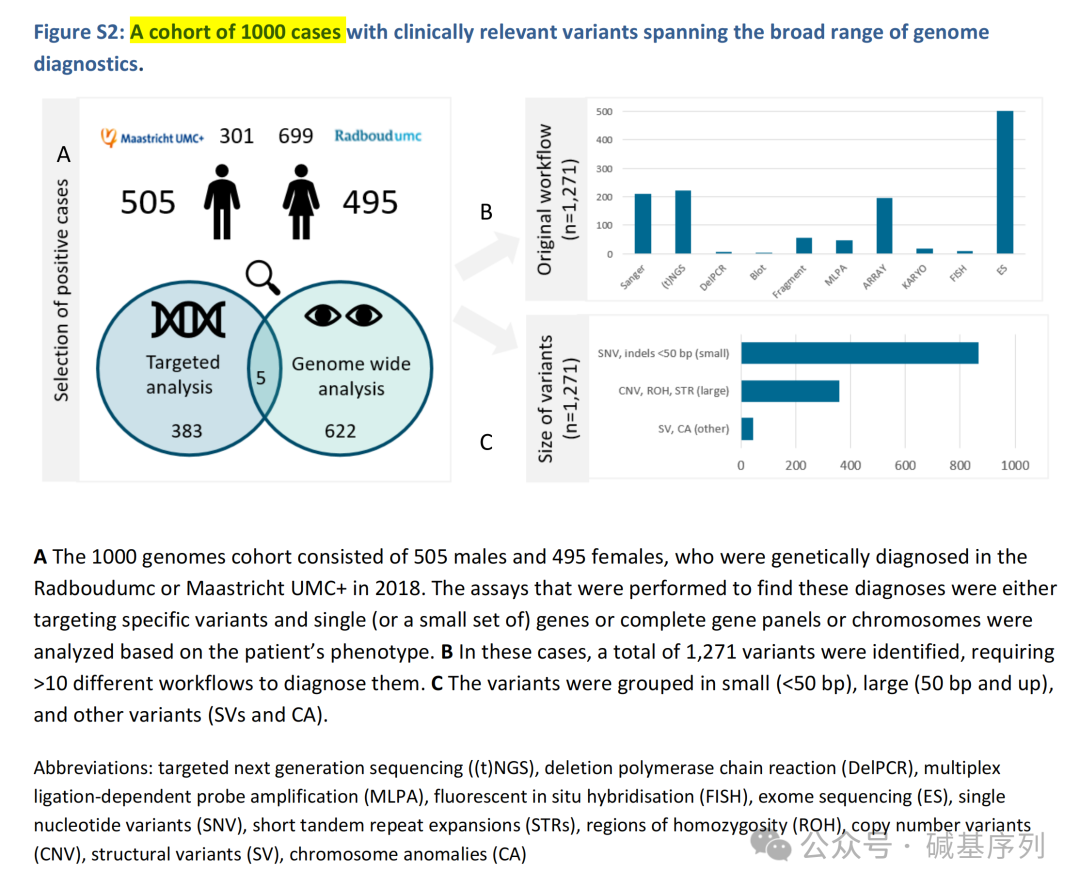
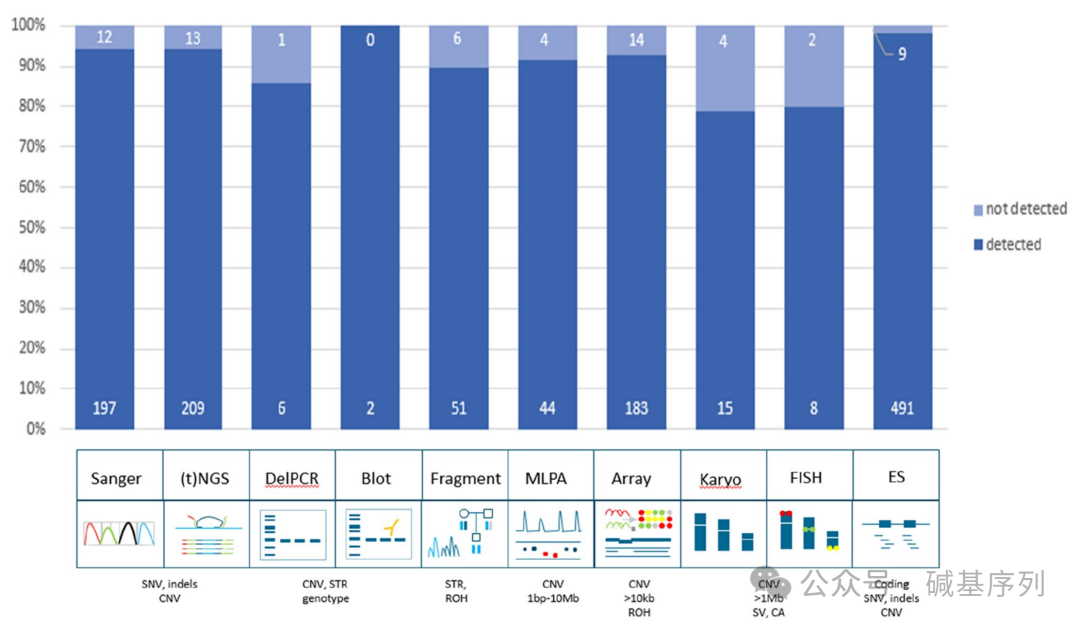
在先前的检测诊断流程中,378个病例使用的是靶向分析,如特定变异位点、单一基因或小基因组;617个病例使用的是基因组检测法(如基因panel或其他全基因组分析);其余5例同时使用两种方法。 共鉴定出1271个与疾病相关的变异,分为三大类:第一类为小的变异(n=860),如SNVs、不超过50个碱基对的indels;第二类为结构变异(n=366),如CNVs、STRs;第三类为其他类型的变异(n=45),如SVs、染色体异常(CAs)。 GS的检出效能
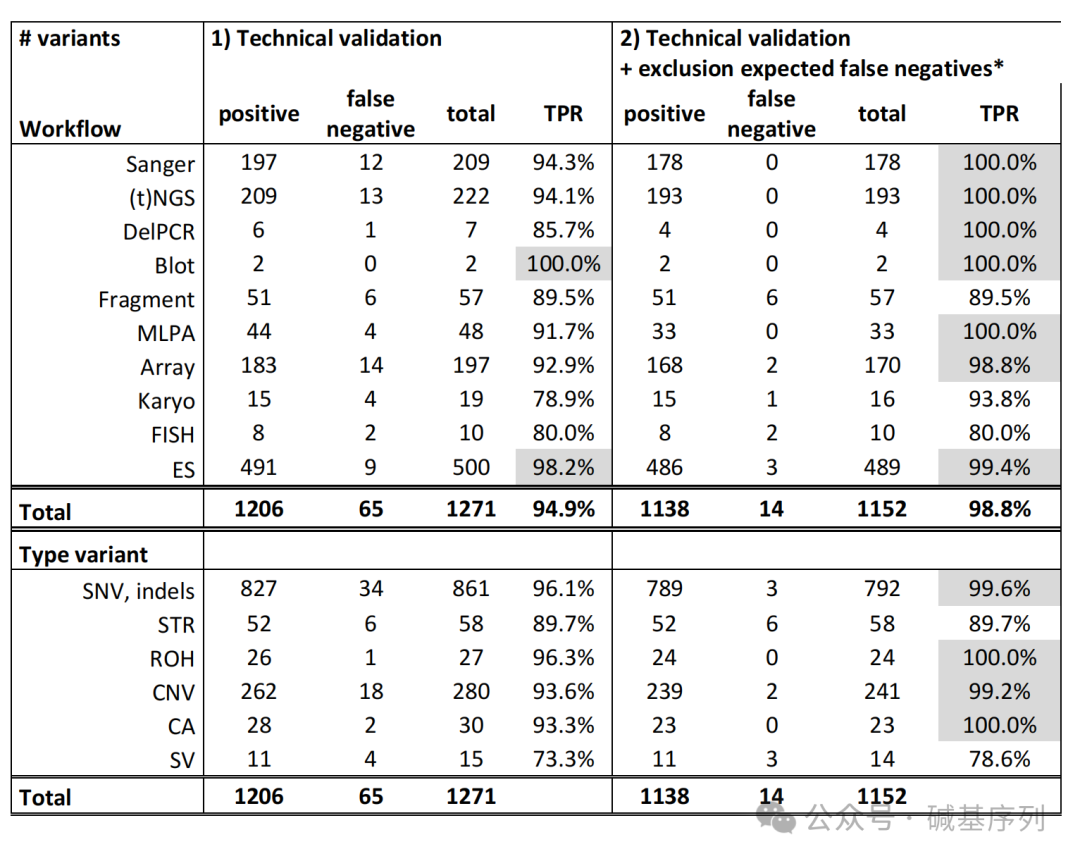
在此研究中,GS能够检出绝大多数变异(94.9%, 1206/1271)。
小的变异(<50bp)检出率为96.1%(826/860);大的变异(123 bp~72.8 Mb)检出率为93.2%(341/366个);其他变异的检出率为86.7%(39/45)。 根据变异的易检出性将其细分为两组:A组为预期容易被GS直接检出的变异(n=1152);B组为预期难以被GS检出的变异(n=119)。A组:检出率98.8%(1138/1152);B组检出率57.1%(68/119)。
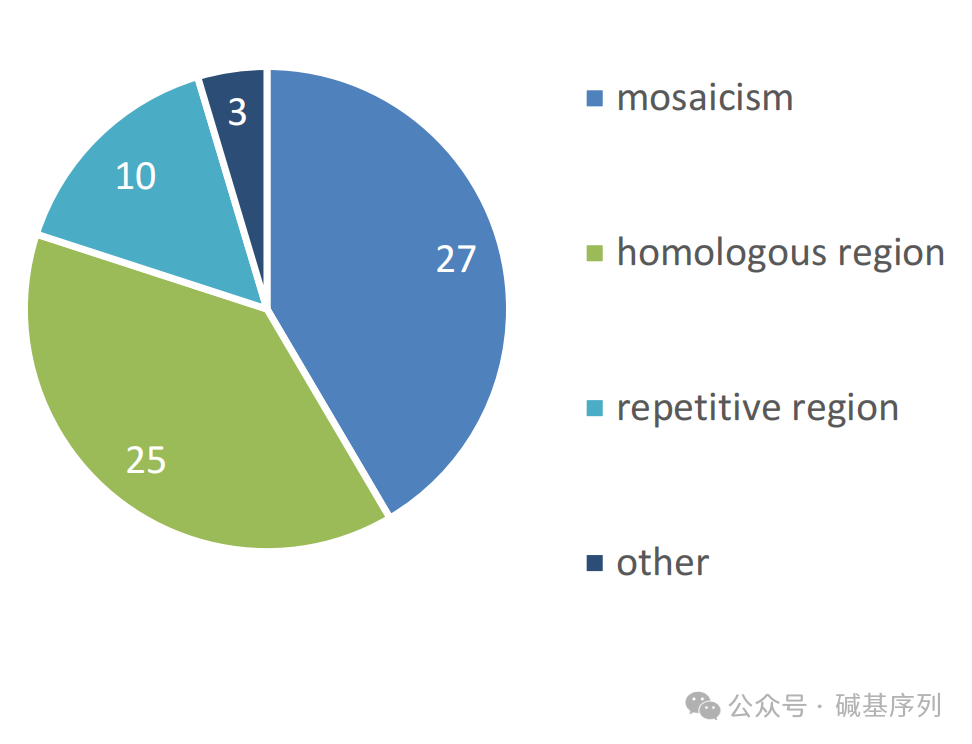
在人工复核后,仍然有65个变异未被检出,分别为嵌合性变异(n=27, 体细胞和线粒体变异)、高同源性区域变异(n=25, 如STRC基因或视蛋白基因家族中的变异)、短串联重复序列或重复序列(n=10, 如FMR1基因和Robertsonian易位)、其他少见类型的变异(n=3)。特别是,小的嵌合性变异,其可检测比例下限为13%。
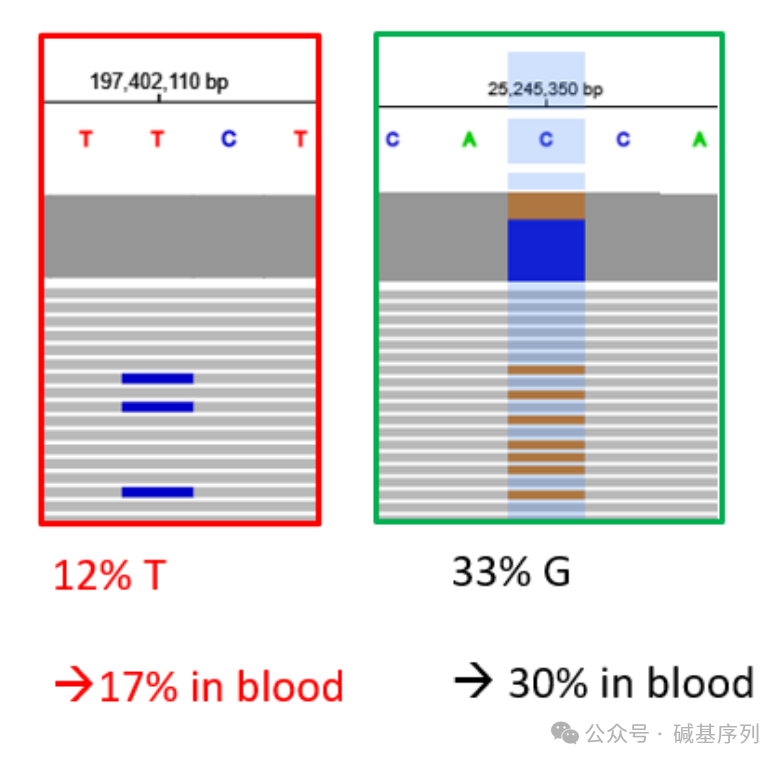
例如:SF3B1基因的嵌合性变异【Chr2(GRCh38):g.197402110T>C】最初是通过靶向NGS方法在血液样本中检出,其比例为17%;虽然GS的50个reads中有6个(12%)reads提示变异的存在,但GS的VCF文件不提示该变异。 例如:KRAS基因的嵌合性变异【Chr12(GRCh38):g.25245350C>G】最初是通过靶向NGS方法在血液样本中检出,其比例为30%;GS的VCF文件能够提示,且比为15/46(33%)。
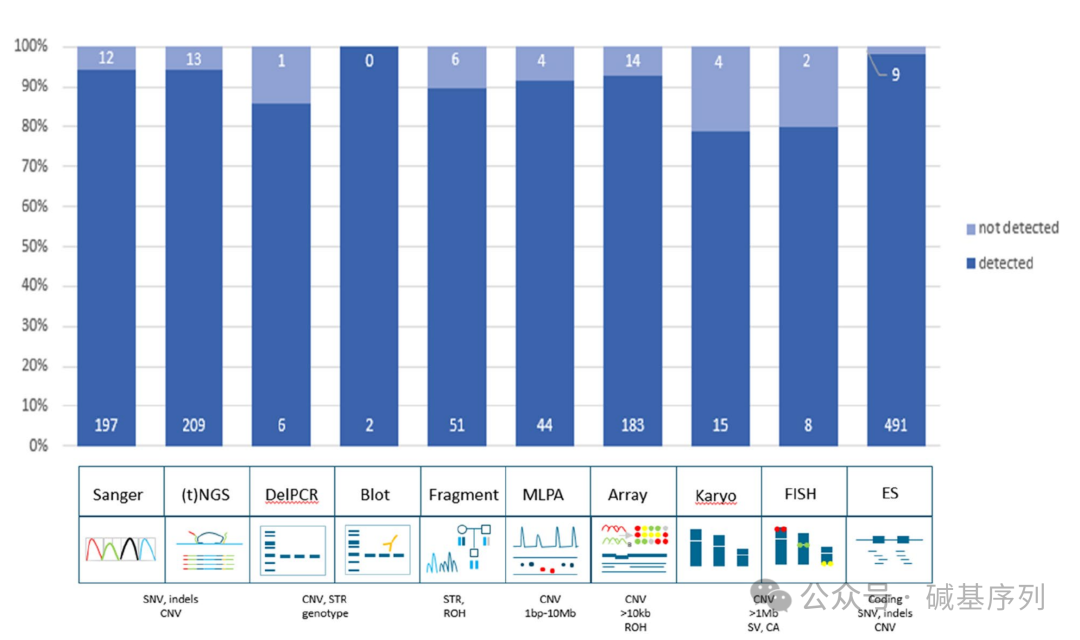
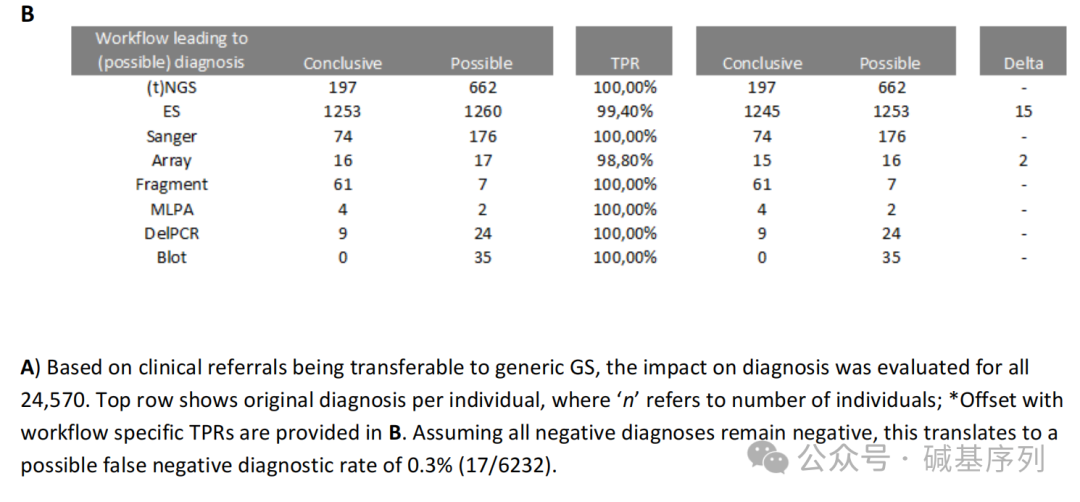
接下来,将1,271个变异重新分配到原来的检测工作流程中,以确定每个检测工作流程检测不同变异类型的总体效能,发现这些检测工作流程的检出效能跨度从染色体核型分析的79%到Southern印迹的100%之间。对每个检测工作流程的TPR进行分析,除重复长度分析、核型分析、FISH外,所有检测工作流程的TPR>为98%。
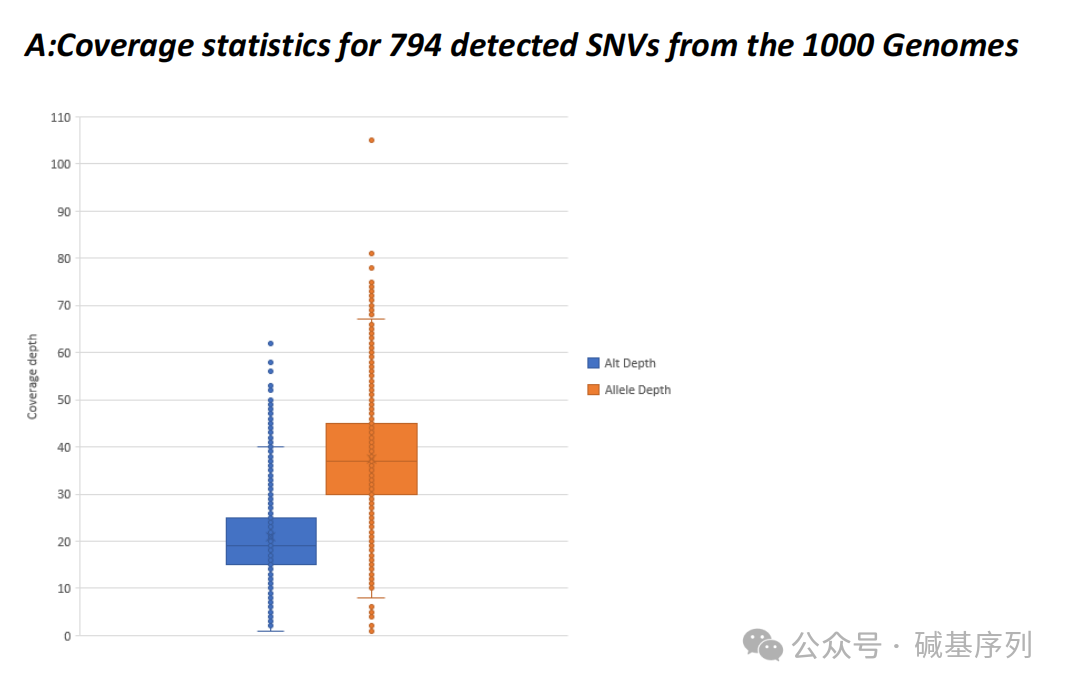
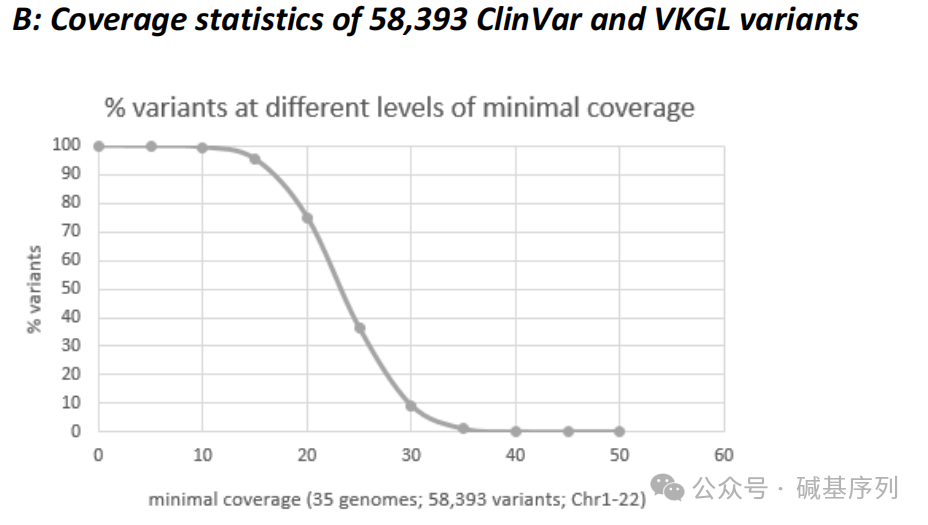
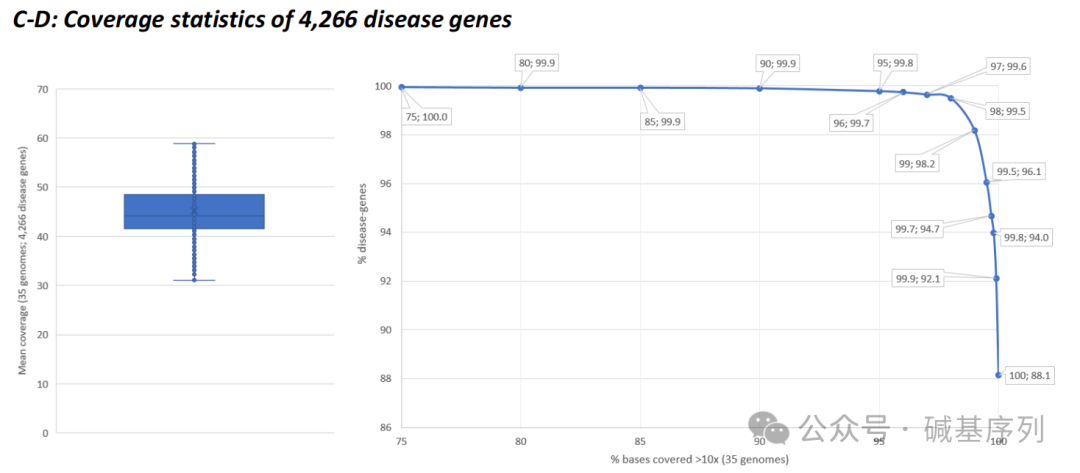
GS测序深度分析
从变异知识库(Variant Knowledge Base, VKGL)、临床变异数据库(ClinVar)中提取了58,393个常染色体显性或隐性遗传疾病相关变异的基因组坐标,分析35个基因组数据集中的这些变异位点,结果显示:99.5%的变异测序深度≥10倍。
GS在常规实践中的应用及其影响 建模分析
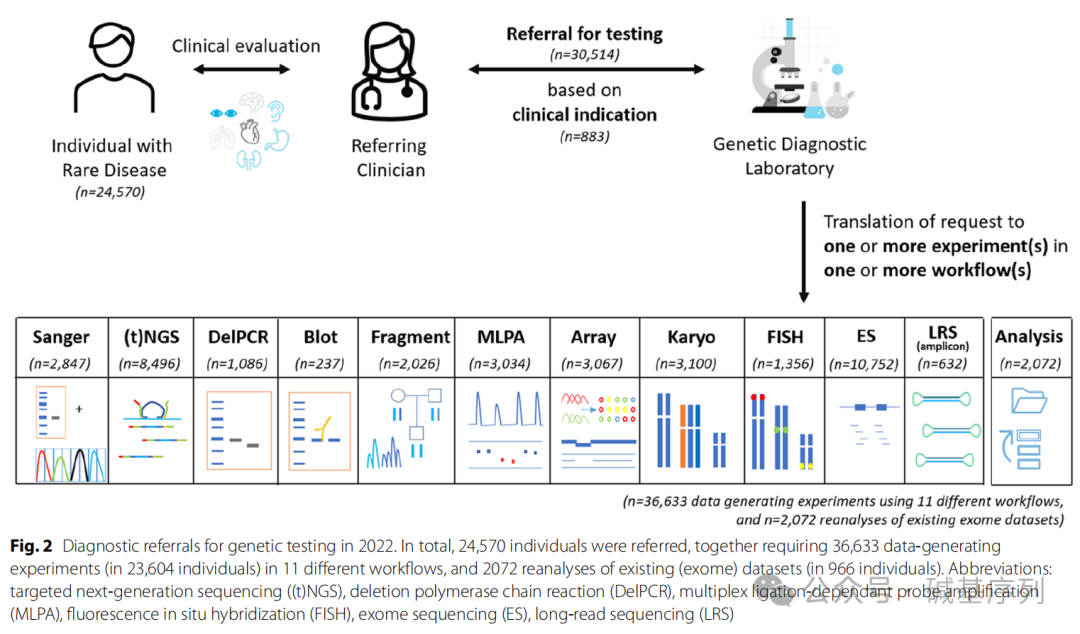
2022年,三级遗传诊断中心共收到30,514例转诊,鉴定24,570名罕见病患者的原发性生殖系DNA变异,共记录了883种不同的转诊原因,其中排名前十的临床指征占所有转诊总数的21%。平均每个患者有1.24次转诊记录,其中82%的患者仅有一次转诊。特别指出的是,对966名患者(2072次转诊)的外显子组测序数据进行重新分析。另外28,442次转诊则通过11种不同的检测工作流程进行了36,633次的湿实验室操作。
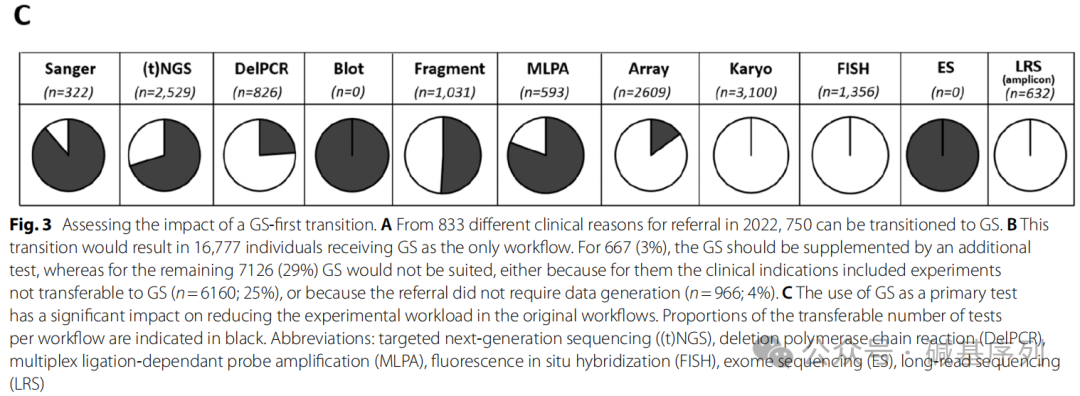
从临床诊断的视角分析,883个转诊原因中有750个(占85%)可以通过GS得到解决。剩余的133个转诊由于各种因素无法通过GS解决,其中体细胞变异(占53%)、同源序列变异(占13%)。
从实验室操作的视角分析,优先进行GS检测不仅能够完全替代WES、所有Southern blot实验,还可以大大减少其他检测工作流程,比如Sanger测序(减少89%)、MLPA(减少80%)、靶向NGS(减少70%)。
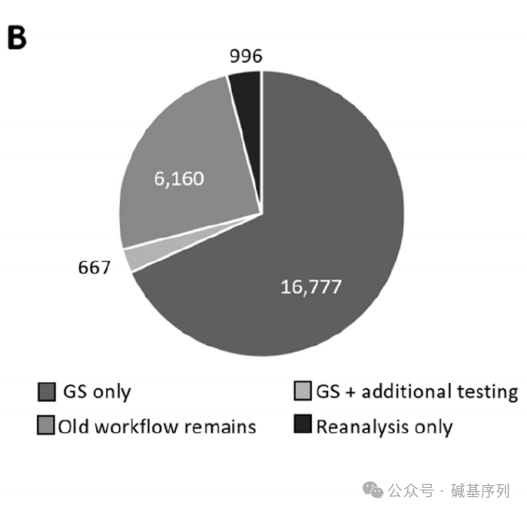
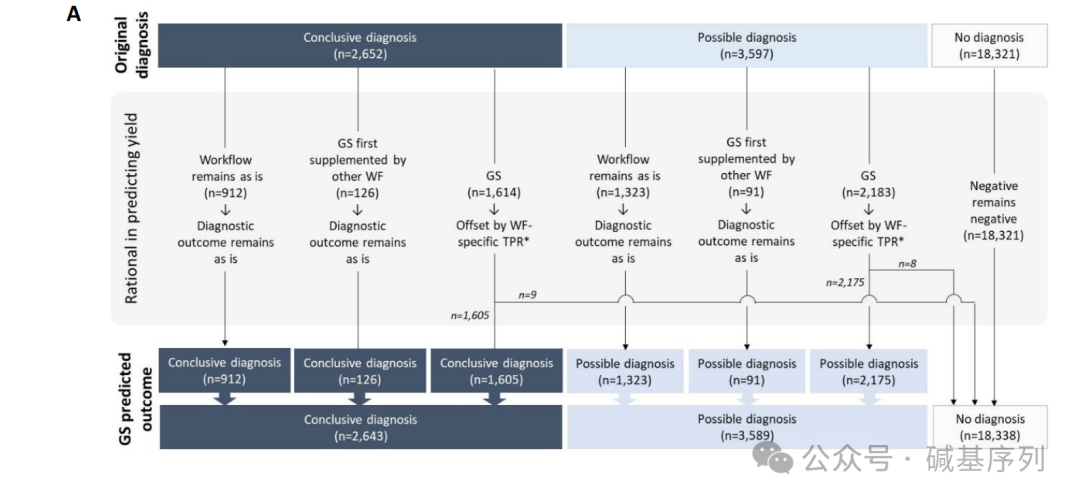
更为重要的是,GS优先检测所有患者的诊断轨迹分析表明,GS可作为16,777名患者(占68%)的首选检测方法。 GS优先策略对总体诊断效能的潜在影响 2022年,总24,570例患者,共6,249例得到结果,其中2,652例患者(占10.79%)获得明确的分子诊断,3,597例患者(占14.64%)获得可能的诊断,18,321例患者未得到诊断。
如果使用GS优先检测,共6,232例患者可以得到结果,其中2,643例患者(占10.76%)可以获得明确的分子诊断,3,589例患者(占14.61%)可以获得可能的诊断。 综合起来,GS优先策略可能会漏诊17例患者(约0.07%,17/24,570)。 讨论总结 在过去的十年中,将GS作为常规检测手段一直存在争论。GS可以在单一实验中识别几乎所有的遗传变异。然而,由于预期的更高的检出率迄今为止并未实现,GS的广泛应用在成本方面受到了阻碍。 因此,本研究重点关注GS作为一种通用诊断罕见病的检测方法,评估GS取代基因诊断实验室现有的全谱系工作流程的潜力,研究发现GS检出了95%以上致病变异,但其检出效能在不同类型的变异和检测方法中存在差异。 在这1000个样本中,通过GS,96.1%小的变异(<50 bp),93.3%大的变异、86.7%其他变异被检出。与ES相比,GS能够更好地检出结构变异,这是GS的一个优势。从概念上讲,这是正确的,因为它在基因组上具有更均匀的覆盖。 通过GS,可以获得的额外诊断,不仅是SVs,而且识别复杂SVs的分辨率通常远远好于其他技术。然而,现在的数据表明,从GS中捕获的SNVs/indels比SVs更完整。无论如何,必须指出,由于纳入标准要求使用EDTA血液中的DNA,因此评估的SVs总数是有限的。 任何技术都有其局限性。在这里,GS未检测到5%(65/1271)的变异。例如,在高度同源区域。测序深度大约30×的短读长GS在检测嵌合性变异时也会遇到困难。然而,从短读长的数据中恢复这些变异的解决方案也是有的:对于嵌合性变异,增加GS测序深度;而生物信息学算法优化有助于检出位于复杂的同源的基因组区域的致病变异。例如,在分析中成功使用了针对SMA和CYP21A2位点的算法,还开发了识别同源区域变异的工具。因此,在本研究中,检出了预期认为可能无法通过30×GS检测的119个变异中的68个。例如,位于高度同源区域基因(STRC和OTOA)的变异、嵌合状态的变异(比例> 14%)。 对于这些变异,一个样本通常需要多个检测实验才能明确。例如,CHD7基因单倍剂量不足引起CHARGE综合征,需要进行Sanger测序和MLPA。而GS可以同时检测SNV/indels、CNV、影响CHD7的SVs。例如,短串联重复引起的疾病,对GS来说具挑战性,因为短读长测序技术可能无法捕获串联重复的全部长度。然而,本研究的数据表明,尽管对于某些重复,GS无法获得确切的长度,但GS能够识别那些重复长度超出正常范围的变异。这种提示性结果可以用其他特定的检测手段进一步确定重复的大小。从检测效能的角度来看,可以认为仍然需要第二个检测工作流程,但可以使得第二个检测工作流程高效起来。 一个实验室是否需要有效地采取GS作为一线检测策略可能取决于实验室的一些特定因素,如实验室的规模,能使用的检测工作流程数量、患者类型。专门进行ES的实验室可以轻松地转向GS。值得注意的是,在大规模实施GS之前,建议进行特定地点的卫生经济影响分析,其中成本效益评估是关键的。在实施GS后,可以减少一些检测工作流程和劳动力,从而减少一些成本,以及临床数据解释相关的成本。 本中心的模型显示,750/883(85%)的转诊可以通过GS检测完成,68%被转诊的患者可以通过单一工作流程和单一实验完成,仅3%需要额外检测,表明对于71%的个体(n=17,444),GS检测优先策略是有益的。有15%的患者不适用于GS,大多数这些患者需要通过核型分析,FISH和/或阵列来检测体细胞结构变异,或者变异位于复杂的基因组区域,这类变异可以通过基于扩增子的长读长测序策略进行评估。 体细胞的SNV/indel可以通过更高深度的测序(如~100~350)来检出。 对于需要通过核型分析、FISH和/或微阵列进行体细胞变异的检测,OGM可以取代这些工作流程,作为GS的第二个主要的通用检测手段,可并行使用,但用于不同的转诊原因。 同样,更通用的长读长的基因组测序可为复杂的基因组区域的变异或变异大小超出短读长可检测范围(例如重复扩展)提供成本效益策略。 对于任何技术解决方案,都需要仔细评估所需的覆盖率,以及与旧技术相比假阴性率的影响。引入新技术需要仔细权衡利弊。对于GS,本研究突出了与实验室检测效率相关的优势,但也表明并非所有先前检测到的(可能)致病的生殖系变异也能从GS中检测出来。 对GS的假阴性率进行客观量化评估,使用先前诊断策略获得的所有诊断作为金标准,发现GS优先策略可能会导至0.3%的假阴性诊断率。虽然这对个体患者来说是不利的,但总体而言,这需要权衡利弊。例如,以牺牲核型分析为代价的基因组微阵列,不能检测明显的平衡性染色体重排不。随着ES替代Sanger测序用于遗传和临床异质性疾病,在获得突变目标大小的同时,也丧失了敏感性。 尽管失去了一些阳性诊断,但仍提高了整体的诊断率。到目前为止,GS的整体诊断优势仍有限。例如,GS在神经发育障碍疾病中的诊断率仅提升了1.3%,在先天性四肢畸形中的诊断率提升了17%。 在细胞遗传学上发现的明显的平衡性染色体重排且是de novo易位和倒位的患者中,大约1/3实际上是基因组不平衡,大约2/3平衡性染色体异常涉及致病机制。随着在GS数据中检测和解释结构变异的经验增长,可以识别出更多的倒位、易位、其他结构变异。与旧的检测工作流程相比,使用GS优先策略将为罕见病患者提供附加价值,从而可能补偿GS所带来的0.3%的诊断假阴性。 最近的前瞻性并行和随机GS研究中表明,比较GS与当前(非GS)的检测流程,可以获得类似的变异类型和诊断率。鉴于此,值得强调的是,即使从分析上提供了全基因组序列,但也可以根据临床要求进行有针对性的基因变异解读。例如,当GS代替Sanger测序时,可以优先考虑单个基因变异,或者分析CNVs。随着对罕见非编码变异与疾病关系的知识增加,以及检测复杂基因组区域变异的生物信息学算法的改进,GS似乎可以作为罕见病诊断的第一线检测手段。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号