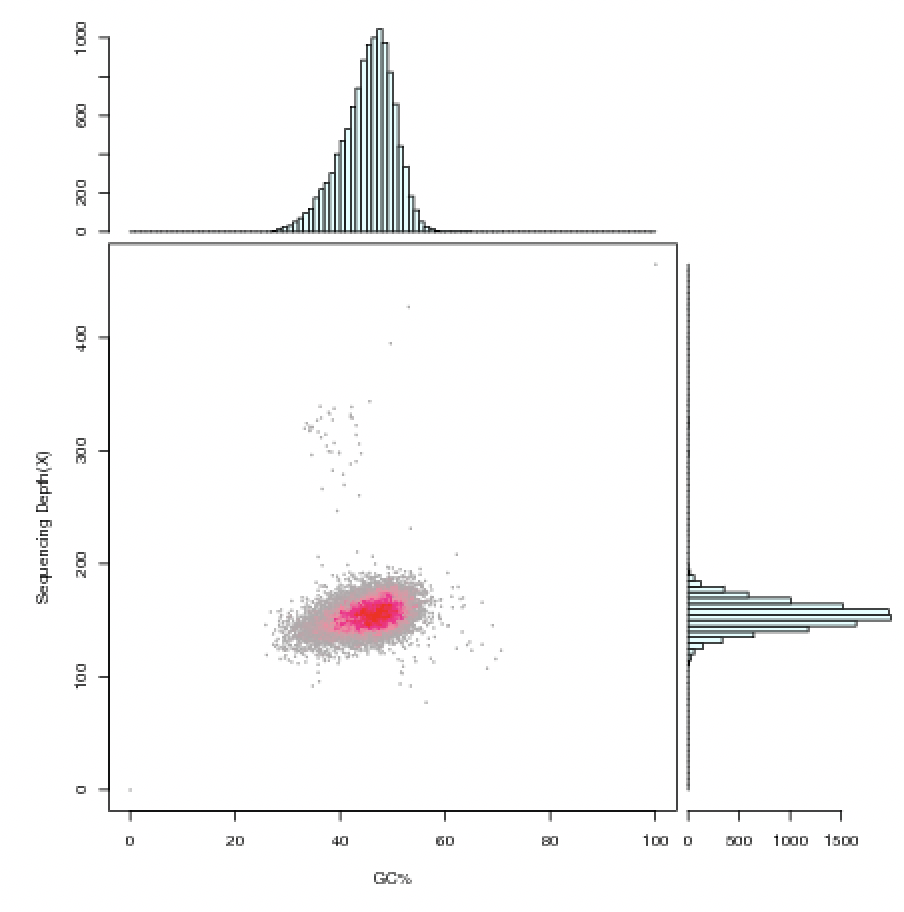
GC-depth 图是表征整个基因组 GC 含量和深度分布的关系,具体方法是对基因组序列进行一定长度切分,每个窗口都有特定的 GC 含量和 Reads 覆盖深度,对应图中的一个点。对于较纯的样本,会集中在某个区域,向四周弥散。而如果 GC-depth 图分开成了多个集中区域,一般意味着该组装结果中包含来自不同来源的 DNA,特别是 GC 层面上如果分开的话,有外源污染可能性很大。GC 不分离,仅深度分离时,也有可能是部分来自质粒的 DNA,需要结合其他信息,如 NT比对结果来具体分析。
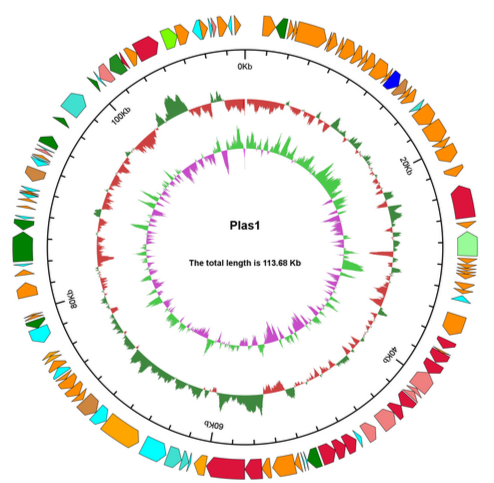
图1 GC 含量与测序深度(Depth)关联分析统计图 组装软件会将测序数据看作来自同一个基因组的前提下进行组装,如果有外源DNA混杂,不同来源的DNA中的序列会对组装产生干扰,为保证组装的准确性,只能将可疑的部分切断成不同的碎片序列,从而导至最终的组装只能拿到碎片化的序列。 我们分析样本基因组的测序深度发现:染色体的reads测序深度在100x左右,成环质粒的测序深度在80x左右,而不成环质粒的仅在20-40x左右。所以,很可能是因为这些样本的质粒拷贝数少,导至质粒的测序深度没有达到足够的乘数,因此质粒组装没有成环。
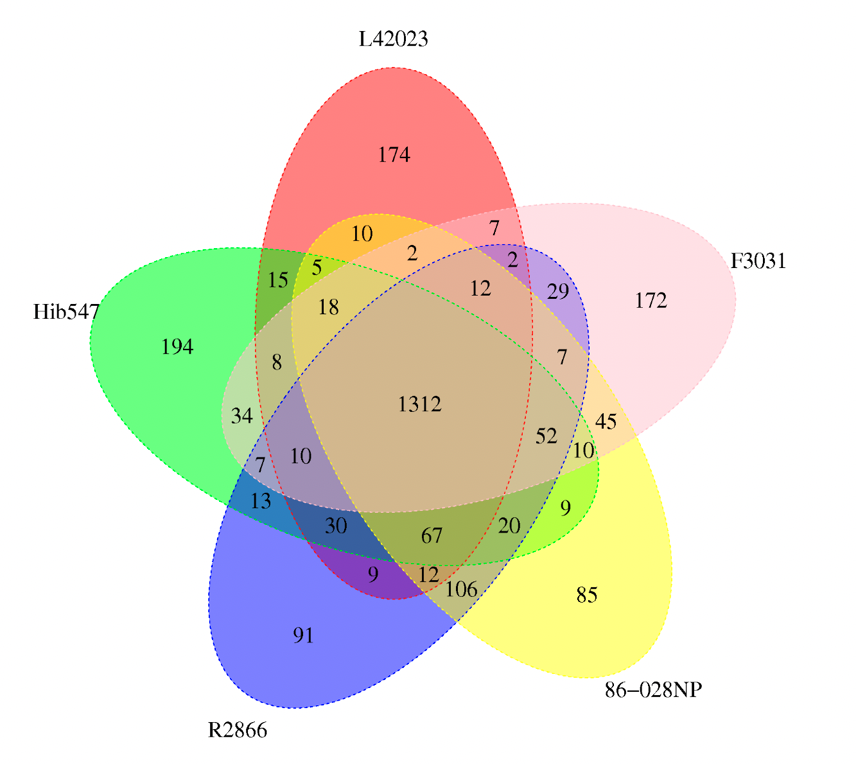
图2 质粒圈图 真菌基因预测有三种方法:从头预测、同源预测及基于转录组数据预测。从头预测使用Augustus软件,同源预测使用Genewise软件。基于同源比对需要提供同种的编码基因序列,而且越近缘对预测结果越好。客户最好能提供近缘物种的编码基因信息,或者组装好的转录本序列文件。三种方法预测的结果将通过EVM进行整合,所以如果客户能提供近缘参考序列和转录数据,结合三种方法的预测结果最好。 组装序列中是否存在此基因?如果不存在,可能是这个基因没有被组装出来,那么肯定也不会被注释到。如果有这个基因,但是没有被注释,有可能数据库中没有这个基因或者是没有被本地数据库收录。 Q:关于ncRNA注释,为什么注释不到 5S/16S/23S 的序列? 在使用denovo方法预测ncRNA序列时,需要完整的ncRNA序列,才能确认ncRNA的结构,而由于ncRNA,特别是16S和23S序列,往往本身就有一定的重复序列成分,在组装过程中很容易组装不完整,如果整条rRNA没有拼接成一条完整序列,是无法预测得到相应的rRNA序列的。如组装较好,该样本对应的物种在数据库注释的少,还是会注释不到。在一些真核新物种的样本中,会经常出现18S等数目为0的情况,这个是因为之前这个物种并没有进行过18S序列测序,所以数据库以及常用软件中没有收录该物种的18S序列,所以没有办法在组装结果中预测出18S。 Identity表示相似性,即序列的一致性。这个值越高,表示同源性越高,序列相似度越高,越有可能是行使相同功能的基因。Score 是比对得分,是打分矩阵计算出来的值,是搜索算法决定的,这个值越大说明你的序列跟目标序列匹配程度越大;Evalue值就是Score值可靠性的评价。它表明在随机的情况下,其它序列与目标序列相似度大于Score值的可能性,所以它的分值越低越好。 韦恩图中每个椭圆表示一个样本,每个区域上的数据表示在且仅在此区域的样本中出现的group的个数,如下图,一个group表示一组具有大于50%相似性、序列长度差异低于0.7的基因集。表格中统计的是基因的个数,图说明的是基因集的个数。
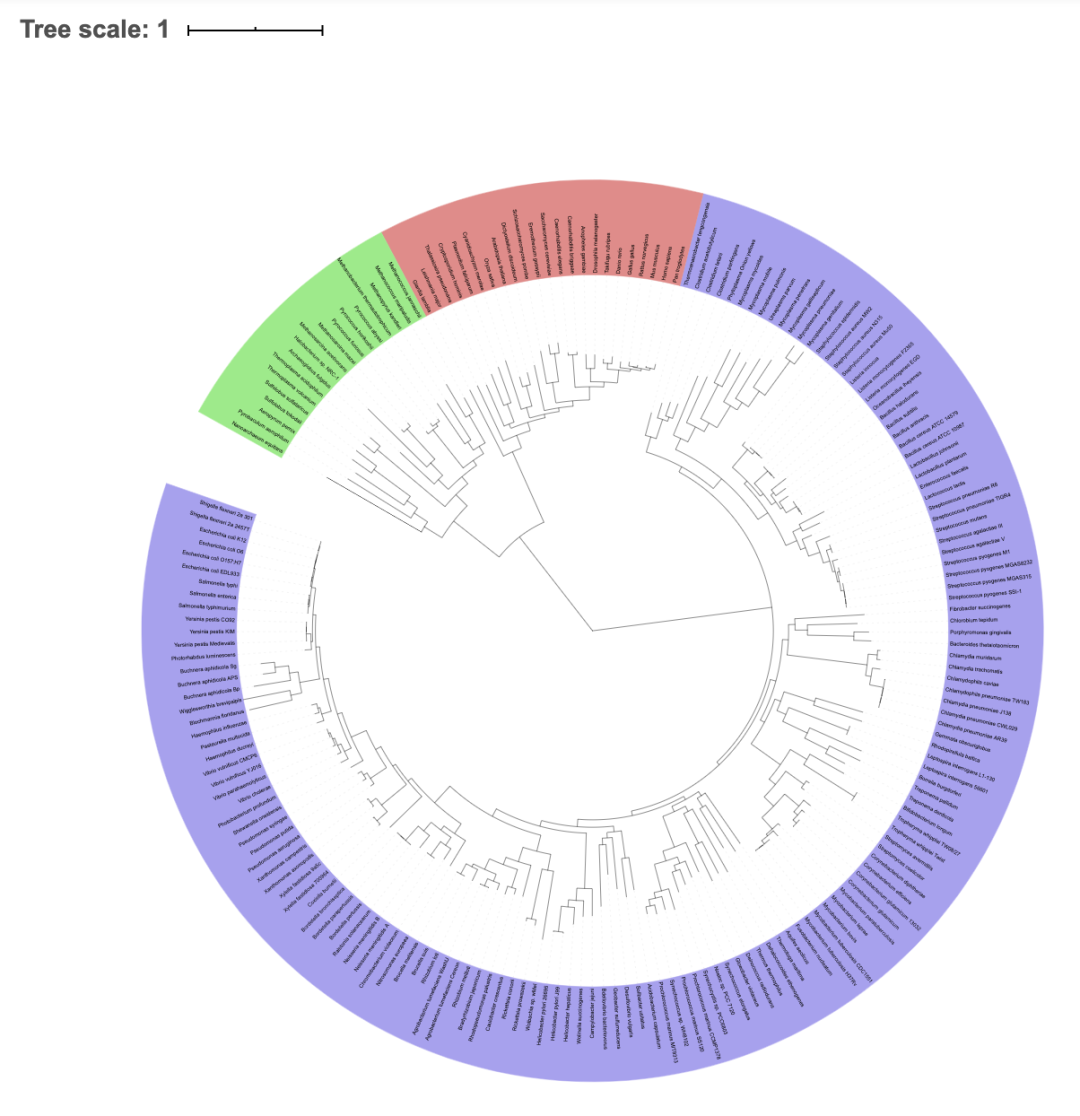
图3 Venn 图 利用MUMmer比对软件,将每个样本与参考序列进行全局比对,找出样本序列与参考序列之间有差异的位点并进行初步过滤,检测出潜在SNP位点;提取参考序列SNP位点两边各100bp的序列,然后使用BLAST软件将提取的序列和组装结果进行比对,验证SNP位点。如果比对的长度小于101bp,则认为是不可信的SNP,将去除;如比对上多次,认为是重复区域的SNP,也将被去除;最后用BLAST、TRF、Repeatmask软件预测参考序列的重复序列区,过滤位于重复区的SNP。最后得到可靠的SNP。 系统进化树的构建有三种方法: 1,基于SNP建树:用样本和参考菌株群体的SNP矩阵构建系统进化树。按照相同顺序将所有SNP相连,获得相同长度的序列,用PhyML软件构建系统进化树。 2,基于core-pan分析建树:用core-pan分析鉴定出样本的单拷贝core基因,利用MUSCLE软件进行蛋白多序列比对,用TreeBeST软件构建系统进化树。 3,基于基因家族建树:用基因家族聚类鉴定出的单拷贝直系同源基因结果,利用MUSCLE软件进行蛋白多序列比对,用TreeBeST软件构建系统进化树。
图4 系统进化树图 看了以上问题的解答后,大家是不是意犹未尽呢?是不是还想了解更多的知识呢?或是想和同行的小伙伴们一起讨论下目前的研究进展?那么就加入我们微生物研究部研讨群吧,群里不仅会有不定时的电子版资料、简书、B站分享,还会有直播喔,扫扫下方二维码,寻找志同道合的朋友们吧~ |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号