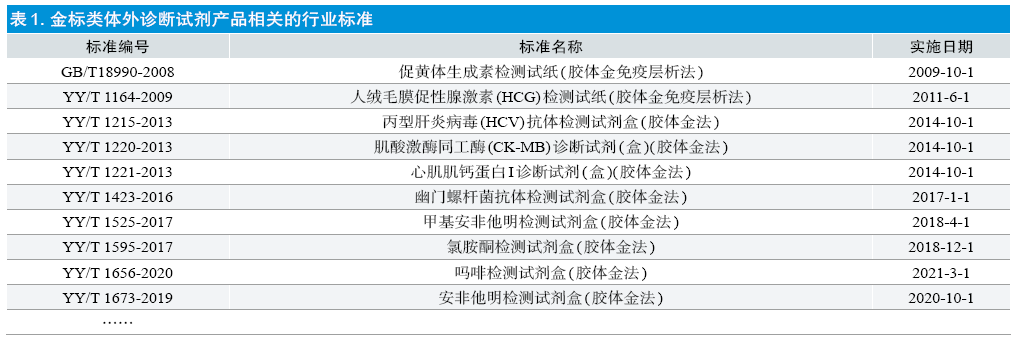
来源:本文刊登于《中国医疗器械信息》杂志2020年第19期 作者:戴清1 张绍千2 张苏琳1 吴猛1 吴浩3 张莉莉1 单位:1 江苏省食品药品监督管理局认证审评中心 (江苏 南京 210002) 2 江苏省药品监督管理局 (江苏 南京 210008) 3 泰州医药高新技术产业园区新药申报服务中心 (江苏 泰州 225300) 内容提要:结合金标类体外诊断试剂本身的特点,从审评、现场检查、研发、生产的角度出发,列举一些在设计开发过程中可能会出现的问题,并针对这些问题进行分析,旨在为企业在具体操作层面应加强的关注点提供技术参考。 关 键 词:体外诊断试剂 胶体金 设计开发 金标类检测试剂是利用胶体金免疫技术,通过竞争法或夹心法,采用胶体金标记的抗原或抗体包被于聚脂膜、玻璃纤维膜或者其他载体,同时把相关抗原或抗体包被在硝酸纤维膜,应用侧向层析法的原理检测待测人体样本中的抗体或抗原的快速检测试剂,属于按医疗器械管理的体外诊断试剂的一种,根据市场需求、不同检测指标的特异性、不同厂家原料及生产工艺的特性等可以设计成为定量产品、半定量产品以及定性产品[1,2]。 产品的质量源于设计,设计开发是探究摸索医疗器械产品的预期用途、原理机制、工艺技术转化、风险分析等的过程,是医疗器械产品实现的一个最为关键的环节,产品安全可靠有效的固有属性是由它决定的。人们常说“产品质量首先是设计出来的,其次是制造出来的,再其次是服务出来的”,反过来说,有设计缺陷的产品,再精心制造、精心服务也于事无补。因此,设计开发过程控制是医疗器械质量管理体系的重要环节[3]。 以下从设计开发部分阶段入手,结合金标类体外诊断试剂的特点,分别阐述其在设计开发各个阶段常见问题及分析和建议。 1.设计开发输入 YY/T 0287-2017 中规定:设计开发输入通常包括:适用的法律法规要求(主要指相关标准、与产品有关的规范或指导原则等);根据预期用途确定的性能、功能以及安全的要求;以前类似的设计所提供的相关信息,如类似产品发生的不良事件等;风险管理的计划、方案输出等等。 企业在设计开发输入过程中,经常会输入不适用的行业标准和指导原则。目前已发布的金标类体外诊断试剂产品相关的行业标准见表1,指导原则见表2,这些标准和指导原则以定性产品为主。大多数定量产品的行业标准和指导原则的适用范围中明确标注不适用于各类胶体金标记试纸等,因此企业要在设计输入时候特别注意自己产品的适用性,需要根据产品的适用性选择合适的标准和指导原则,避免在产品注册审评阶段被提出发补因未选择适用的行标或指导原则而造成的返工,如重新型式检验甚至重新进行临床试验或临床评价等。
除以上标准之外,建议企业重点关注通用法律法规的输入,尤其要特别关注国家局最新发布的有关法规。经常部分企业会出现法律法规输入不全的问题,如缺少近两年发布的《医疗器械注册质量管理体系核查指南》《医疗器械生产企业供应商审核指南》《医疗器械不良事件监测与再评价管理办法》《YY/T 0316-2016 医疗器械风险管理对医疗器械的应用》《YY/T 0466.1-2016 医疗器械 用于医疗器械标签、标记和提供信息的符号 第1 部分:通用要求》等。 2.设计开发输出 设计开发输出应当满足输入的要求,通常包括:①采购信息,如生物原材料(抗原抗体等)、硝酸纤维素膜、胶体金、聚酯膜、玻璃纤维膜、吸水纸等技术要求;②生产和服务所需的信息,如工艺流程图、作业指导书、工艺配方、环境要求等;③技术要求;④原材料、半成品、成品等的检验规程或指导书;⑤规定产品的安全和正常使用所必需的产品特性,如说明书、标签要求等,注意说明书应当与注册申报与批准的一致;⑥标识和可追溯性要求;⑦提交给注册审批部门的文件,诸如综述资料、原材料研究资料、生产工艺研究资料、分析性能评估资料、稳定性研究资料、产品技术要求、注册检验报告、临床评价资料(如有)等。 对于胶体金类体外诊断试剂来说,研发的过程可能使用了各供应商甚至是本公司的合作单位等送来的各种小样,导致设计开发输出时弱化了对于采购信息的要求,忽略了对供应商的审核等,甚至没有签订明确关键指标要求的质量协议或采购合同。例如有的原材料是从母公司定向采购,或直接由母公司赠送而来,拿来就直接使用,忽略了输出诸如原材料抗原抗体的效价、点膜喷金仪器的要求、硝酸纤维素膜孔径、聚酯膜等亲水疏水性等原料关键指标的要求。医疗器械质量管理体系环环相扣,设计开发输出对原材料的质量技术要求一环出现了问题,就会引起其他环节出现连锁反应,最终可能导至产品不合格。建议企业多多关注体系的合规性,不论过程复杂或简单,法规要求的环节都需要不折不扣地完成,不能随意删减。 3.设计开发转换 设计转换是指从医疗器械样品试制、小批量试产、中试到实现质量稳定地、持续有序地批量化、规模化生产的过程。 经常有企业对设计转换的过程不够重视,还是以研发思维来指导生产,研发模式不能及时转换为生产模式。研发是小试,而且经常是同一个人从头到尾完成的,系统误差稳定,小试的试验结果很好,但是中试大试可能会不稳定,对金标类体外诊断试剂来说,胶体金的制备,点膜溶液、标记抗体溶液、样品垫处理液等的配制工艺,不确定因素很多,都有可能会很容易引起这种不稳定。在这种情况下,更应该及时建立标准操作规程,严格控制生产过程。有的企业从生产记录表格的设计中就可以看出生产过程控制的好坏。例如有的配制作为生产过程的关键工序,记录上只有诸如“量取1234.56μL”这些简单的字眼,甚至有的生产记录里连量取多少体积都没有写,也没有计算过程,只有配方比例,生产操作人员的计算过程是另外在随手拿来的本子或纸上完成的,完全不受控。这些操作对于研发人员来说可能很简单,但是对于一线的生产人员来说仅凭这些描述就想保持产品性能的稳定性,保持变异系数(cv)在合适的范围内,可能就很难了。而且有的记录上不能体现一人操作一人复核的生产质量控制过程,如没有操作人员和复核人员的签名等。对于体外诊断试剂来说,很多都是微量的量取,误差可接受范围很小,应该更加注意操作的严谨性和规范性。 设计转换后应更加认识到生产批记录的重要性,需要保证生产过程可追溯。有些企业的生产记录中的物料平衡计算流于形式,如领料某某溶液72.3μL,量取使用了12.5μL,还剩59.8μL,这个59.8μL 是直接把前两个数字相减得出来的。长此以往,最终也不知道到底该溶液具体实际有多少,而且造成整个生产过程无法追溯,如果最终产品质量失控,无法找出是哪个环节出现问题,找不出问题所在。 胶体金类体外诊断试剂生产工艺简单,但是要注意的细节很多,越简单的东西越不能掉以轻心,才有可能在质量管理体系运行的前提下持续生产出性能稳定、合格的产品。 4.设计开发评审 设计开发评审是对设计和开发阶段结果和最终结果的适宜性、充分性、有效性、是否达到规定目标所进行的系统评估活动。评审活动既可以评价设计和开发结果是否能够满足顾客要求、法律法规要求和企业附加要求,也有助于发现设计和开发不同阶段问题并采取有效地纠正预防措施,避免产品设计缺陷和产品不合格对后续发开和生产、生产后活动的不良影响。 评审在设计开发的各个该阶段可能出现的问题有:①重评审过程,轻评审记录。在研发的过程中,有很多过程都需要评审,而研发人员在研发的过程中必然也是进行了这个过程的,否则最后不可能输出一个合格的产品。但是研发过程纷繁而复杂,可能有很多实验在同时进行。研发人员可能最后只简单的记录一下实验结果,评审过程一带而过,而没有按照设计开发程序文件的要求合规的记录评审记录,并进行分析,记录后续如何必要措施,包括纠正预防措施;②记录不规范。为了完成评审记录,很多个评审活动在同一天进行,有的评审记录的表格设计的非常完善,但是填写内容的时候敷衍粗糙,不重视留痕,有的甚至留空,没有填写意见只有各相关人员的签名及日期。评审记录是评审活动的真实反映,不能为了记录而记录,应让记录为以后的设计开发活动服务,起到它应有的作用。 建议企业在评审过程中真实记录自己所做的所有活动,不能有侥幸心理,认为个人或某些人知道就行,记录简单一点。但是人的记忆时间长了可能会出错,记录下来的东西却不会,并且记录是活动的证据,证据需要妥善留存。 5.设计开发变更 研发活动是一个不断试错的过程,很多次试验才有可能一次成功,所以从设计开发输入到输出一气呵成、一次完成的可能性比较小,因此设计开发变更的存在几乎是必然的。 设计开发变更可能发生在设计和开发过程的任何阶段:如输入后、开发中、输出前、输出后,评审、验证、确认、风险分析活动后或产品取得上市许可前或产品上市后。如有的企业一直以来使用的抗体(点膜抗体和/ 或标记抗体)效价突然达不到要求了,造成产品不能顺利通过出厂检验,此时可能需要变更原材料供应商。需要注意的是,根据体外诊断试剂注册管理办法、医疗器械生产监督管理办法、医疗器械说明书和标签管理规定等相关法规的规定,已取得上市许可的医疗器械在实施设计开发更改后,需按照法规规定履行变更注册申请、说明书备案、生产条件重大变更报告等义务,获得批准后方可将更改后的产品投放市场。已注册的三类、二类体外诊断试剂,医疗器械注册证及其附件载明的内容发生变化,注册人应当向原注册部门申请注册变更,并按照相关要求提交申报资料。 以二类的胶体金类体外诊断试剂为例,其注册证的附件为技术要求和说明书,以下如:①抗原、抗体等主要原材料供应商发生变化;②阳性判断值或者参考区间、检测条件发生变化;③注册产品技术要求中性能指标和/ 或检验方法发生变化;④储存条件或者产品有效期发生;⑤包装规格、适用机型(厂家和型号)发生变化;⑥预期用途发生变化,如增加临床适应症、增加临床测定用样本类型等;⑦可能影响产品安全性、有效性的其他变更,需要申请许可事项变更。不同的变更事项需要根据不同的要求提供相应的评估资料,内容上需要符合2014 年44 号公告的要求。同时需要根据企业受控的程序文件的要求对设计开发变更进行控制,留下完整记录,并进行风险管理。部分企业在变更注册过程中提交的资料从形式上来说都不完整,如:未提交原产品技术要求复印件、原产品说明书原件、变更后产品说明书只有一份等。 建议企业应当多了解不同的变更类型需要提交什么样的评价资料,不敷衍、不偷工减料,这样能减少变更注册需要的时间。需要注意的是,如果在技术审评发补的过程中引起了产品某些变化,也需要进入设计变更程序。 6.小结 本文结合金标类体外诊断试剂本身的特点,依据法律法规,从审评、现场检查、研发、生产的角度出发,通过对设计开发输入、输出、转换、评审、变更环节的常见问题列举并进行分析,希望能为企业在具体操作层面应加强的关注点提供技术参考。 参考文献略 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号