更多详细信息还得再等几天
“这速度,很可以了” 相信大家都看到了世和大panel在FDA获批的新闻。
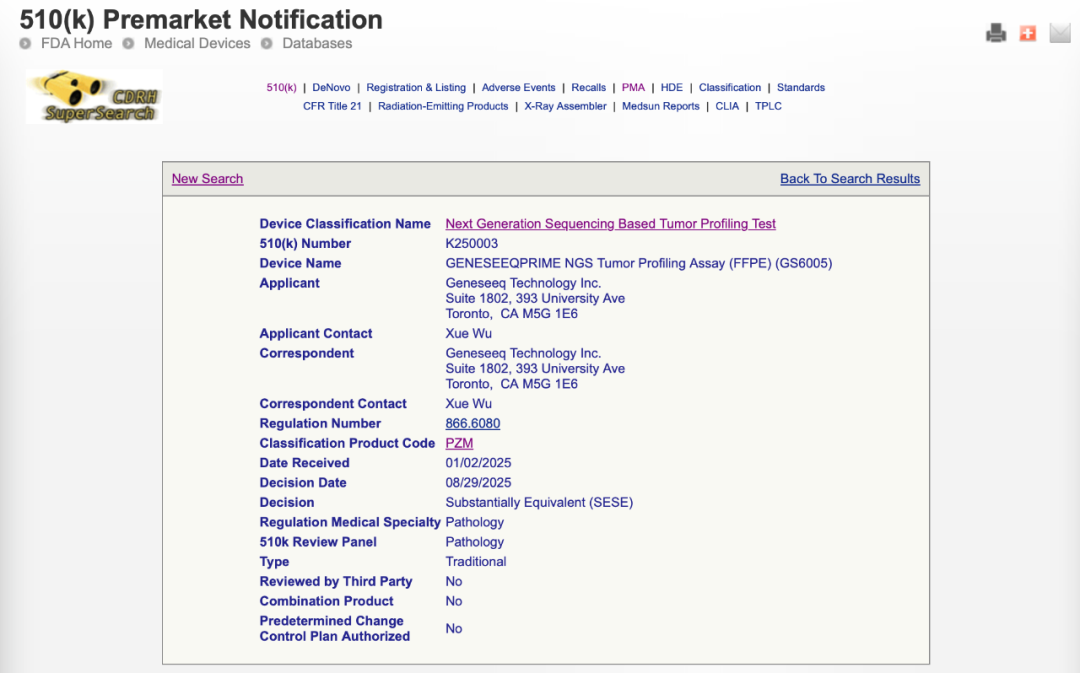
恭喜世和,是个好消息。 有不少朋友好奇这个产品在FDA是怎么获批的,借着官方信息我们简单聊两句。 1. 510k,II类 从FDA的官方信息来看,Geneseeq PRIME是走的510k审批路径,设备类型是Next Generation Sequencing Based Tumor Profiling Test。
和PMA路径的最大区别是,510k大概率不涉及到伴随诊断,仅做性能验证。 其预期用途是一个定性的体外诊断检测: 可以理解成425个基因(包括MSI+TMB)都是NMPA定义下的二级位点——或者NMPA没有定义过的三级位点。
这确实是中美审批原则上的巨大差异,NMPA既不接受肿瘤NGS试剂盒作为二类,也不会接受肿瘤NGS试剂盒不包含一级位点(不管是伴随诊断、补充诊断还是辅助诊断)。 走510k这条路径中我们最耳熟能详的产品是MSKCC-IMPACT,也是首个获批的大panel。 510k的审批逻辑是找一个“可比产品(predicate device)”,证明申报产品与这个已经获批的产品“等效”(substantially equivalent)。
世和的官方消息说PRIME不是single-site而是试剂盒,所以我们盲猜是选了下面这个产品来对比 PGDx elio,505基因(包括了MSI+TMB),试剂盒 *这个试剂盒也是510k路径获批,当年选的predicate device是MSK-IMPACT
最后的谜底可以等到FDA公开Decision Summary的时候揭晓——类似于CMDE的审评报告。 一般会在获批后3-6周发布~我们持续关注。 2. 中美双报 这个试剂盒对世和最大的帮助是承接药企合作的生意——全球多中心临床。 理论上可以做到一个试剂盒同步完成一款药物的中美伴随诊断。 考虑到Foundation One和Guardant 360在国内无法获批,Geneseeq prime确实可以说是当前的独一份。 当然要迈过的坎也不少,比如: 510k路径下获批的NGS试剂盒目前还没有成功添加CDx label的先例、PRIME如何在美国本土F1CDx的“垄断”和G360 tissue的“崛起”下完成商业化布局,以及是否能够为国内的TMB试剂盒增加CDx label。 但这都是后话,路总是一步一步走的,走了第一步才能去想后两步。 3. 加鸡腿 510k的申请一般分几个阶段: 受理审查(Acceptance review)、实质性审查(Substantive Reive)、最终审查(Interactive review) 虽然FDA的承诺/目标是90个工作日左右(大约120天)完成审查,但这个过程中免不了会遇到FDA要求补充信息。 所以通常6-9个月的时间是需要的。 以前面提到的PGDx elio为例,从accept到最终获批花了大约9个月。
世和的PRIME递交是今年的1月2号,获批是8月29号,总共不到8个月。
这注册效率,是真不错。 这鸡腿,至少得加两。
|


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号