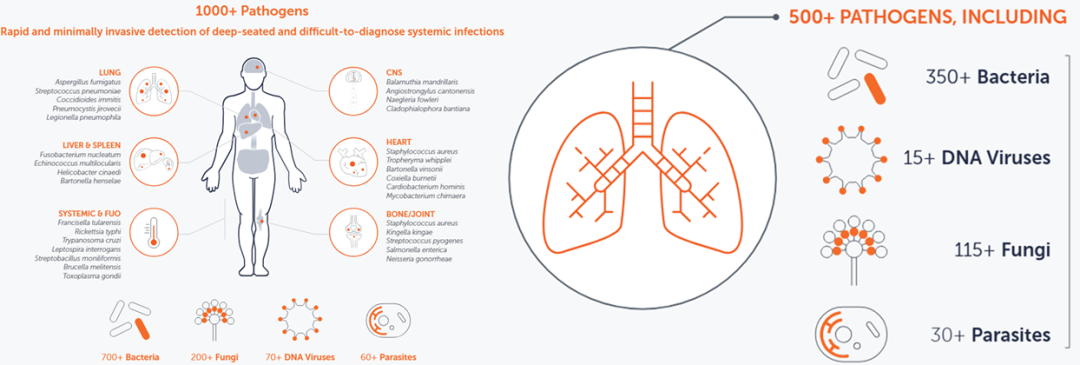
近日,广东省佛山市报告的基孔肯雅热疫情,让这种由伊蚊传播、以高热、皮疹和剧烈关节疼痛(甚至导至患者弯腰行走)为特征的急性传染病,进入公众视野。 基孔肯雅病毒(CHIKV)最早于1952年在坦桑尼亚被发现,主要流行于热带和亚热带地区。 然而,此次疫情也引发了一个关键问题:面对基孔肯雅热这类相对少见甚至罕见的病原体,临床医生如何能快速锁定“真凶”,为精准诊疗提供明确的病原学依据? 这正是病原宏基因组测序(mNGS)技术大显身手的领域! 本系列文章(共两期)将带您了解,从数据库的维度mNGS如何成为发现罕见或新发病原体的强大工具。 mNGS的核心优势在于其“无偏性”——它能一次性检测样本中几乎所有微生物的核酸信息。 但要最大化发挥这一价值,关键在于两大环节的协同优化:
既往,技术优化的关注点多聚焦于湿实验的提升。而本期,我们将重点探讨:如何将mNGS的“知识库”——病原体基因组数据库做得更“大”、更“强”,从而为精准识别罕见病原体奠定坚实基础。 病原mNGS的“大”数据库,其根基在于整合全球权威的微生物基因组资源(如全球微生物数据中心WDCM、综合微生物基因组数据库IMG,以及抗生素抗性基因库CARD、毒力因子库VFDB等)。 但这绝非简单的数据堆砌!关键在于“去粗取精”:经过严格的筛选和整合,最终构建一个高质量、特征明确的“临床级”病原体参考数据库 这个“大”数据库的核心价值在于其“广度”与“质量”:
数据库不仅要“大”(覆盖广),更要“强”(质量高、注释准) FDA-ARGOS 数据库树立了很好的示范。
Karius 的“强”数据库实践 Karius(成立于2014年)的核心产品——Karius Test,正是依托其强大的独家病原体基因组数据库,结合NGS与AI分析,实现对病原体的高效检测。
它们的“强”体现在:
如同升级了“渔网”的网眼密度和覆盖范围,一个做“大”的数据库,能让我们捕获到更多样、更稀有的病原体“踪迹”。 国内病原mNGS技术的蓬勃发展始于2016年前后,众多团队的成立加速了技术的迭代与优化。其无偏性检测、广谱覆盖的核心价值,也迅速获得了临床权威指南的认可:
数据库建设:规模竞赛与质量基石 作为mNGS技术的核心“知识库”,病原体基因组数据库的建设至关重要。其核心理念早在2017年美国病理学家协会(CAP) 团队发表的奠基性文献《Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection》中就已明确:物种全面性 (Comprehensiveness)、注释准确性 (Annotation Accuracy)、序列完整性 (Sequence Completeness)。
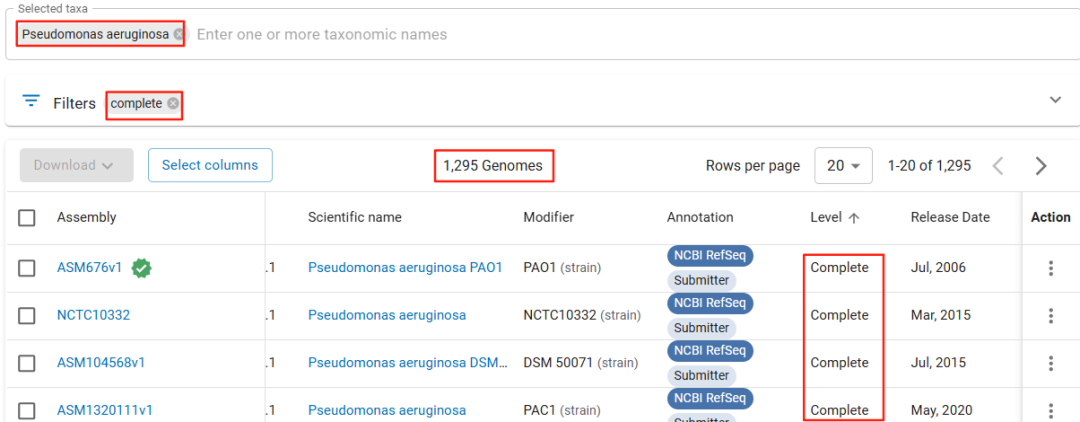
数据库的“大”与“强”,如同车之双轮、鸟之两翼,共同推动着mNGS从“能检出”走向“检得准、读得懂”,深刻改变着感染性疾病的病原诊断格局。 二、 宏基因组数据库做“强”——“Pan”genome,升级“渔网”的材质 数据库仅仅“大”(物种覆盖广)还不够,更要“强”——能精准识别病原体内部的关键差异。这就引出一个核心挑战: 难题:单一参考基因组的“盲区” 当我们检索一个常见病原体(如铜绿假单胞菌 Pseudomonas aeruginosa)的基因组时,NCBI上仅“完整”级别的基因组就有上千个!这些基因组大小不一、序列各异。关键问题来了:选择哪个版本作为参考才是“正确”的?
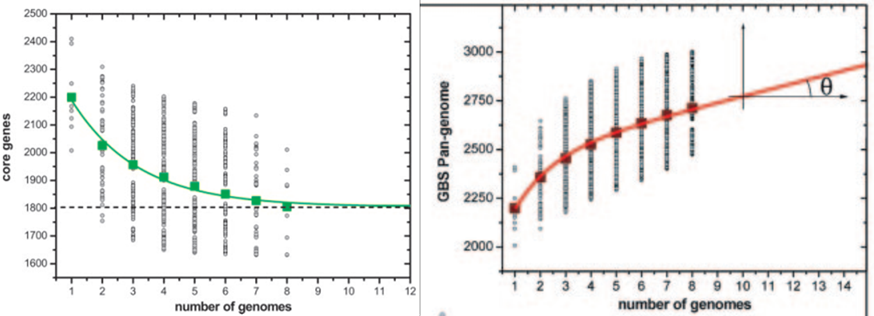
破局:泛基因组(Pan-genome)的“兼容” 2005年,一项发表于顶级期刊《PNAS》的研究提出了革命性的微生物“泛基因组(Pan-genome)” 概念,为上述难题提供了创新的解决方案。它将一个物种的全部基因划分为:
泛基因组 (Pan-genome) = 核心基因组 + 非必需基因组 它代表了一个物种完整的基因“宇宙”,是一个动态的、不断扩充的基因库。 理解泛基因组,是解析病原体致病机制、进化历程和制定精准干预策略的基础。 “做强”数据库的关键:构建泛基因组 将“泛基因组”理念融入病原体基因组数据库建设,意味着:
这就如同将“渔网”的材质从普通纤维升级为高精度复合材料:网眼(代表检测分辨率)并未变小(物种覆盖未缩减),但网线的“灵敏度”和“特异性”极大提升,能更精准地捕获病原体内那些决定致病性、耐药性的关键“小鱼”,从而实现真正意义上的“精准识别”与“深度解读”。 这就是数据库“做强”的核心内涵之一。 “知道泛基因组是什么,但它如何真正提升病原mNGS的临床检测效能?又将面临哪些挑战?”,我们下一期揭晓。。。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号