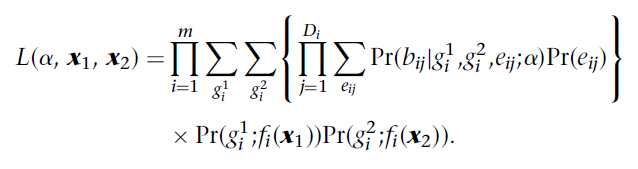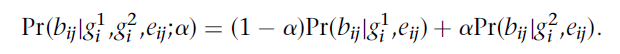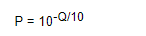本篇主要和大家分享有关NGS检测体细胞突变时判断DNA污染的方法。 一、 FDA产品- MSK-IMPACT的 DNA污染控制方法 1) 判断样本交叉污染(sample mix-up) 计算参考样本和待查样本间的“不一致比例”,即在参考样本中纯合基因型的位点在待查样本中变成另一个等位基因的纯合基因型的占比。此占比在tumor样本和它们对应的匹配的正常样本间一般应为<5%。相反,和其它个体来源的样本间此占比应高,一般为~25%。具有>5%比例的来自同一个体的配对样本和具有<5%比例的来自不同个体的2个样本被标记成交叉污染的样本组。 2) 判断样本污染 计算次等位位点频率,例如有10个SNP位点,则会有20个染色体,如果均为纯合基因型则次等位位点频率为0%,但有其它样本混入时,如果次等位位点的频率超过2%时,此样本就被列入再评估样本中。 3) 判断配对normal样本中是否存在tumor样本 normal样本不应有常见的已知的SNVs和indels突变。首先,在normal样本中检测已知的热点突变位点,如果一个已知的肿瘤特意的突变位点(例如,BRAF V600E)检测突变频率> 1%时,该normal样本就被列入再评估样本中且有可能从下游分析中排除掉此样本。 二、 判断污染的软件 判断污染的软件根据其目的分为2种,一种是只是检测某个样本是否被其它样本污染,另一种是在判断待查样本是否污染的同时,也能找出污染源样本。以下只介绍一种属于第一类的软件verifyBamID。verifyBamID[1]软件可通过输入一个或多个样本的bam文件,输出样本中的污染比例。该软件的算法如下公式1所示:
公式(1) α:污染比例 x1, x2 :分别为待查样本和污染源样本的遗传谱系坐标 SNP位点个数:总共m个,i∈{1,2,3…m} g1i :待查样本的基因型 g2i :污染源样本的基因型 Di :支持第i个SNP位点的reads数,j∈{1,2,3…Di} eij :表示在SNP位点的位置是否发生测序错误的随机变量,发生错误则为1,未发生错误则为0;eij∈{0,1} bij :覆盖第i个SNP位点的第j条染色体上的在该SNP位点的位置处的碱基类型,R代表reference allele, A代表alternate allele,O代表有别于R和A的碱基类型,bij∈{R,A,O} fi(x) :优化过的第i个SNP位点的人群频率 2.1公式1的逻辑 在指定基因型的条件下(Pr(g1i ;fi(x1))Pr(g2i ;fi(x2))),计算某个SNP位点的所有reads上的碱基类型出现的概率乘积,之后计算所有SNP位点的此概率,将所有这些概率的乘积作为计算α的似然值,通过算法Nelder-Mead使公式1进行收敛,即同时解出α和x1, x2的最佳值。 2.2 Pr(bij|g1i ,g2i ,eij;α) 公式(1)中概率Pr(bij|g1i ,g2i ,eij;α)的计算方法如公式(2)所示,即等于在待查样本中看到各SNP位置处出现各碱基类型的概率和污染源样本中看到这些碱基类型的概率之和。
公式(2) 公式(2)中概率Pr(bij|gi ,eij)的计算方法如下表1所示,在指定的基因型条件和已知的base quality的情况下我们可以推测出各位点处碱基出现的概率。例如,比如一个样本的基因型为RR且未发生测序错误时(eij=0),那么支持这个位点的reads上对应位置处应该出现和RR一样的碱基类型R的概率为100%。当发生测序错误时,那么这个位置上会出现有别于R的碱基类型,因此出现R的概率为0,在剩余3种碱基类型出现和alt(A)碱基一样的碱基类型的概率为1/3,出现有别于R和A的的碱基类型O的概率为2/3。 表1 条件概率Pr(bij|gi ,eij)的计算表
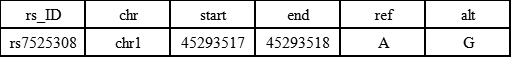
注: a:RR代表纯合野生基因型, RA代表杂合型, AA代表纯合突变基因型 b: O代表有别于R和A的碱基类型,假设有4种碱基类型 2.3 Pr(g1i ;fi(x1))Pr(g2i ;fi(x2)) 公式(1)中Pr(g1i ;fi(x1))和Pr(g2i ;fi(x2))分别代表的是待查样本和污染源样本中在i点处的出现各基因型的概率。在推测出人群频率fi(x)条件下,某个样本的基因型的概率符合二项分布,即gi ~ Binomial(2, fi),因此可根据fi(x)计算出上述2个概率。 2.4 示例 如下表2所示,一个SNP位点的位点名称为rs7525308,在染色体上的位置为45293517(0-based),与参考基因组一样的碱基即ref碱基类型为A,有别于参考基因组的碱基即alt碱基为G。 表2 SNP位点示例
从表3中可知支持该位点的reads总共有10条(见第四列),每条reads上在该位点处的碱基如表3中的第5列信息其中“G”代表和ref不一样的碱基类型,“.”代表和ref一样的碱基类型,第6列表示每个位置处的碱基的碱基质量值。 表3 覆盖rs7525308位点的reads的示例
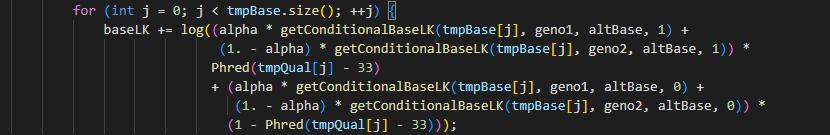
2.4.1计算Pr(bij|g1i ,g2i ,eij;α)和Pr(eij)值 我们从表3知道第5列各碱基类型出现的概率的乘积即为公式1中大括号内的概率。为了得到此概率,我们首选计算第一个碱基G出现的概率即i=0,j=0时的Pr(bij|g1i ,g2i ,eij;α)和Pr(eij)。前者概率根据公式2计算获得;后者表示该位点的base quality的phred score值。 在第一条read的base为碱基G,该位点的alt碱基为G,假定alpha= 0.0775、第一个样本和第二个样本的基因型分别为RR时、此外该位点的base quality为’>’时LK计算脚本如下所示:
上述脚本中getconditionalBaseLK的算法见表1,比如当基因型为RR且eij=1时,bij上看到G的概率为1/3。Phred(tmpQual[j]-33)中将此位点处的质量值“>”转换成phred值的意思,先将此符号转换成ASCII值,之后减去33后即获得Q-Score值,之后按照以下公式(3)获得测序错误率为0.001258925。因此,公式(1)中大括号内的在RR条件下就等于以下公式中的baseLK值。
公式(3) baseLK=log{{[(0.0775*(1/3)+(1-0.0775)*(1/3)]* 0.001258925 + (0.0775*(1/3)+(1-0.0775)*(1/3)]*0.001258925+{[(0.0775*(0)+(1-0.0775)*(0)]*(1-0.001258925)+(0.0775*(0)+(1-0.0775)*(1/3)]*(1-0.001258925)]}} 2.4.2计算Pr(g1i ;fi(x1))Pr(g2i ;fi(x2))值 根据fi值计算gi1和gi2,因gi~Binomial(2,fi),Pr(g1i ;fi(x1))Pr(g2i ;fi(x2))的值为:
AF:fi(x1)或fi(x2)值 GF[0]:当基因型为RR时的概率 GF[1]:当基因型为RA时的概率 GF[2]:当基因型为AA时的概率 参考文献: [1] Fan Zhang. Ancestry-agnostic estimation of DNA sample contamination from sequence reads. Genome Research 30:185–194. |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号