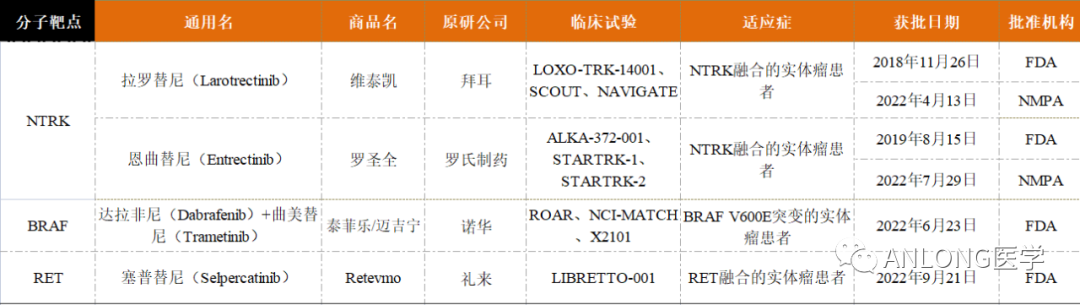
随着“精准医学”时代的到来,肿瘤检测开启了以基因或标志物为适应症的新疗法。目前全球已批准多个生物标志物用于泛实体瘤,包括靶向生物标志物NTRK、BRAF、RET,免疫标志物MSI-H/dMMR、TMB,还有新兴的潜在分子标志物FGFR,开启了肿瘤治疗的新篇章。 靶向生物标志物 国内外已获批实体瘤药物的分子靶点有NTRK、BRAF和RET,相关药物及获批信息见下表。
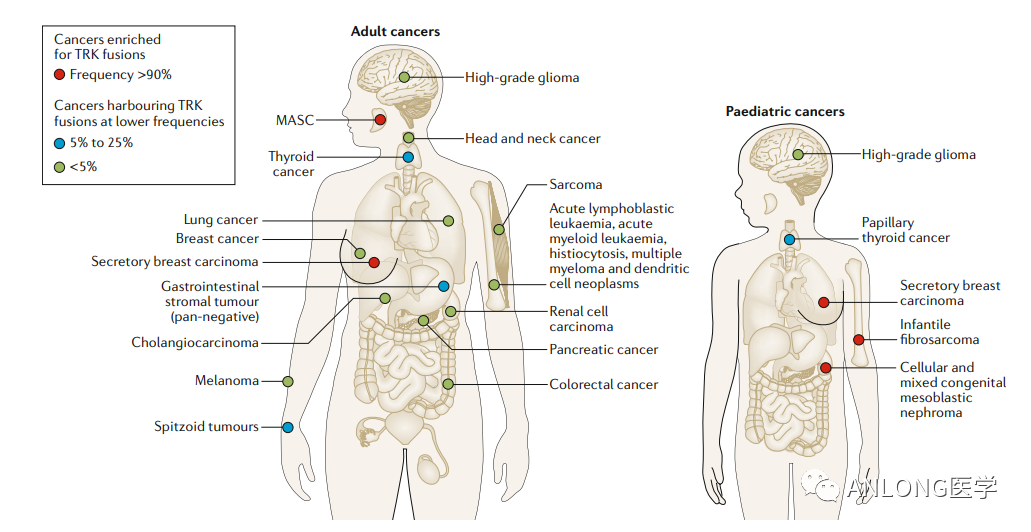
获批实体瘤靶向药物汇总 NTRK NTRK基因包含NTRK1、NTRK2 和 NTRK3,分别编码原肌球蛋白受体激酶(TRK)家族TRKA、TRKB和TRKC三种蛋白。若染色体内或染色体间发生重排导至NTRK基因家族与其它基因发生融合,便会使TRK蛋白的结构性激活,驱动肿瘤的发生。目前已发现NTRK融合存在于超过45类癌种中,在常见肿瘤类型中的发生率较低,如肺癌(0.2%)和结直肠癌(0.3%);但在罕见的肿瘤中较为常见,如婴儿纤维肉瘤(90.6%)、分泌性乳腺癌(92.9%)、分泌性唾液腺癌(79.7%)、先天性中胚层肾癌(21.5%)等。
NTRK融合在成人和儿童肿瘤中的分布和发生频率 《中国实体瘤NTRK融合基因临床诊疗专家共识》强烈推荐所有晚期成人和儿童实体瘤患者均进行NTRK融合基因检测,并且根据不同癌种的特性采取不同的检测策略。目前NTRK基因融合的检测方式包括免疫组化(IHC)、荧光原位杂交(FISH)、逆转录聚合酶链式反应(RT-PCR)和二代测序(NGS)等。2022年两大NTRK抑制剂拉罗替尼、恩曲替尼纷纷在中国获批上市,开启泛癌种靶向治疗新时代。 1 拉罗替尼 2022年4月13日,NMPA批准拜耳的拉罗替尼(Larotrectinib,维泰凯)上市,用于治疗符合下列条件的成人和儿童实体瘤患者:
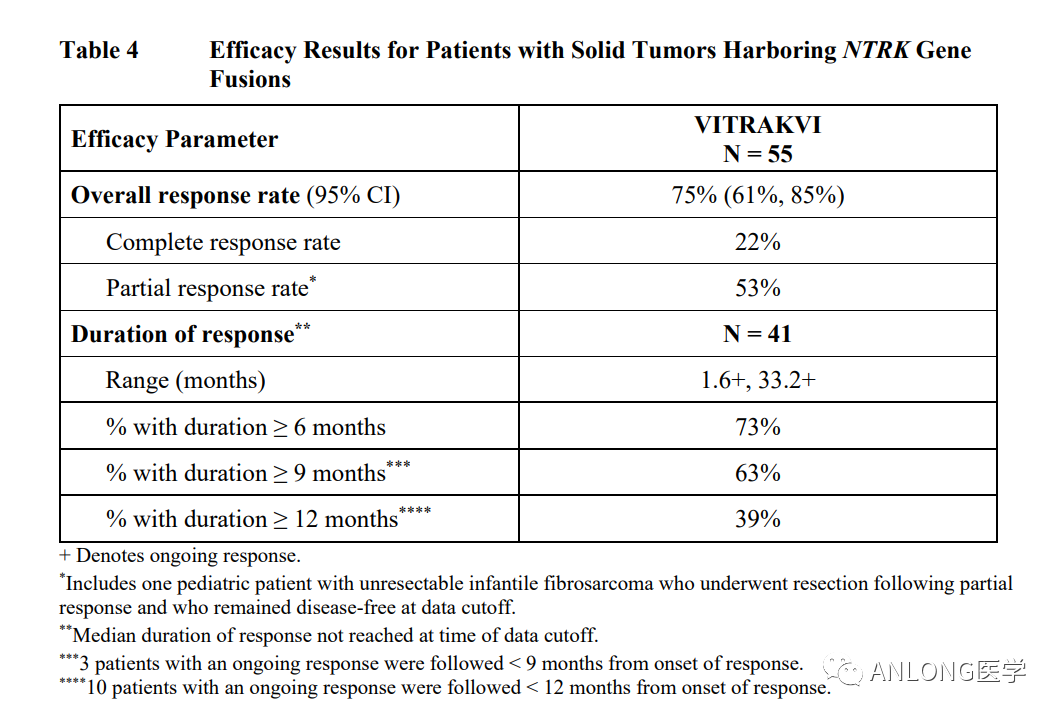
早在2018年11月26日,FDA已批准拉罗替尼上市,用于治疗携带NTRK基因融合的泛实体瘤成人和儿童患者。这是第一个获得FDA批准的不分癌种、只看突变的广谱抗癌靶向药,并被证明在儿童和成人的17种肿瘤中都有效。 拉罗替尼的获批是基于三项大型临床试验LOXO-TRK-14001(NCT02122913)、SCOUT(NCT02637687)和NAVIGATE(NCT02576431)。研究结果显示,拉罗替尼的客观缓解率(ORR)为75%,其中22%的患者获得了完全缓解(CR),53%的患者获得了部分缓解(PR)。
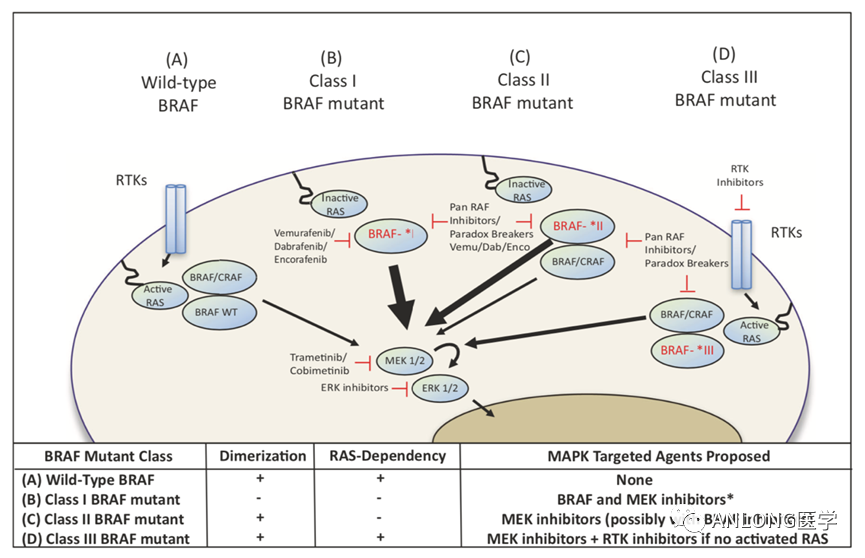
拉罗替尼三项临床试验结果汇总 2 恩曲替尼 2022年7月29日,罗氏制药的恩曲替尼(Entrectinib,罗圣全)获得NMPA正式批准,用于经充分验证的检测方法诊断为携带NTRK融合基因且不包括已知获得性耐药突变,患有局部晚期、转移性疾病或手术切除可能导至严重并发症,以及无满意替代治疗或既往治疗失败的成人和12岁及以上儿童实体瘤患者。早在2019年8月15日,FDA就已批准恩曲替尼上市,用于治疗NTRK基因融合阳性的晚期复发实体瘤成人和儿童患者。 恩曲替尼是一款靶向NTRK和ROS1基因的酪氨酸激酶抑制剂(TKI),具有中枢神经系统(CNS)活性,能够穿透血脑屏障,是临床上唯一一种被证明针对原发性和转移性脑疾病具有疗效的TRK抑制剂,可阻断ROS1和NTRK激酶活性,并导至ROS1或NTRK基因融合的癌细胞死亡。 恩曲替尼的获批是基于三项多中心、单臂、开放性临床试验ALKA-372-001(EudraCT 2012-000148-88)、STARTRK-1(NCT02097810)、STARTRK-2(NCT02568267),试验的主要终点为独立中心审查委员会(BICR)根据RECIST v1.1标准评估的ORR和中位缓解持续时间(DOR),次要终点包括CNS转移患者的ORR和DOR。对NTRK融合基因阳性实体瘤成人患者进行整合分析数据显示,无论基线脑转移状态,成人患者的ORR达到61.3%,DOR高达20.0个月。肉瘤、非小细胞肺癌、涎腺分泌性癌、乳腺癌、神经内分泌癌的ORR也分别高达59.4%、64.5%、84.6%、66.7%和40%。 BRAF BRAF基因位于7号染色体长臂(7q34),是一种重要的原癌基因,编码丝氨酸/苏氨酸特异性激酶,参与调控细胞生长、分化和凋亡等生物学事件。BRAF基因突变是一种广谱的基因突变,在恶性黑色素瘤中发生概率为40%~60%、甲状腺乳头状癌为30%~80%、结直肠癌为5%~15%、肺癌为2%~4%、恶性胶质瘤为3%,并在多种发病率较低的肿瘤包括浆液性卵巢癌、胆管癌、胰腺癌均有一定表达。 根据活化的机制不同,BRAF突变体有三种类型,BRAFⅠ类突变(BRAF V600 D/E/K/R/M 突变),可以强烈激活BRAF激酶活性,组成性活化MAPK通路,且不依赖于RAS作为单体发出信号;BRAFⅡ类突变 ( K601、L597、G464 和 G469等),可作为 RAS 非依赖性二聚体激活下游通路;BRAFⅢ类突变(G466、N581、D594 和 D596等),激酶活性低或缺乏激酶活性,这类突变体依赖RAS并且对依赖ERK反馈的RAS敏感,通过增强其与RAS的结合激活ERK ,并需要分子共存机制使肿瘤中的RAS活化才能起作用。BRAF基因中最常见为V600E突变,占所有肿瘤BRAF突变的95%以上。
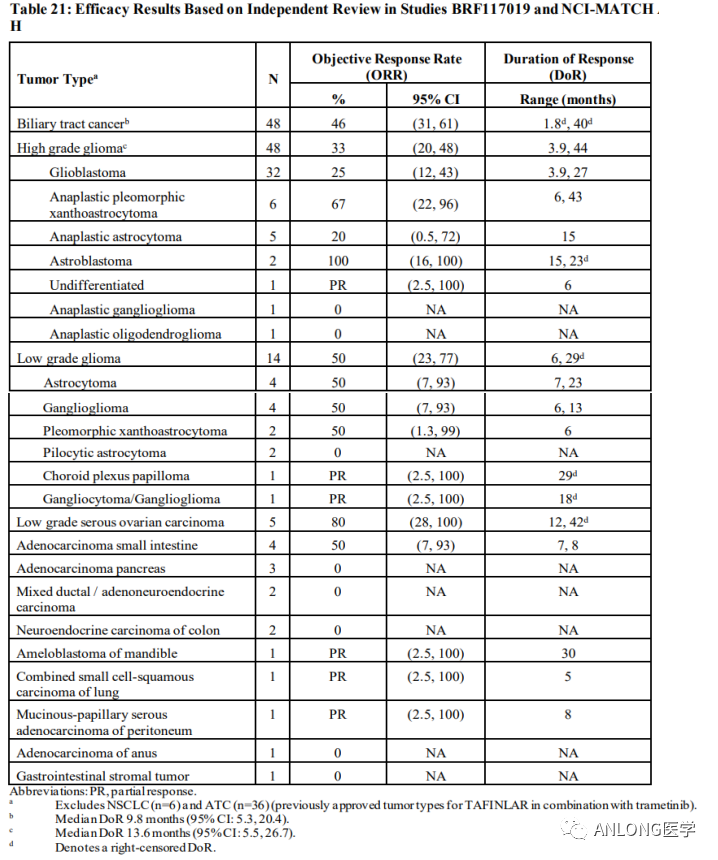
BRAF突变类型 2022年6月23日,FDA加速批准诺华的达拉非尼(Dabrafenib,泰菲乐)联合曲美替尼(Trametinib,迈吉宁)用于携带BRAF V600E突变、不可切除或转移性实体瘤的6岁及以上儿童和成人患者,这些患者在先前的治疗后出现进展且没有其他治疗方案。 达拉非尼和曲美替尼均为小分子TKI类药物,达拉非尼能够结合发生突变的BRAF V600E/K/D基因编码的蛋白产物,发挥抑癌作用。而曲美替尼抑制的位点则是BRAF突变激活的下游基因MEK1/2,两者联合可以进一步放大药物疗效。 本次获批是基于3项临床试验中观察到的临床疗效和安全性数据,包括BRF117019(NCT02034110)、NCI-MATCH(NCT02465060)和一项儿童研究X2101(NCT02124772)。 BRF117019和NCI-MATCH的H组研究共纳入131名成人患者,包括24种肿瘤类型,结果显示达拉非尼+曲美替尼在携带BRAF V600E的实体瘤患者中ORR高达80%,胆道癌患者的ORR为46%,DOR为9.8个月;高级别胶质瘤患者的ORR为33%,中位DOR为13.6个月;低级别胶质瘤患者的ORR为50%,DOR范围为6个月至29个月。
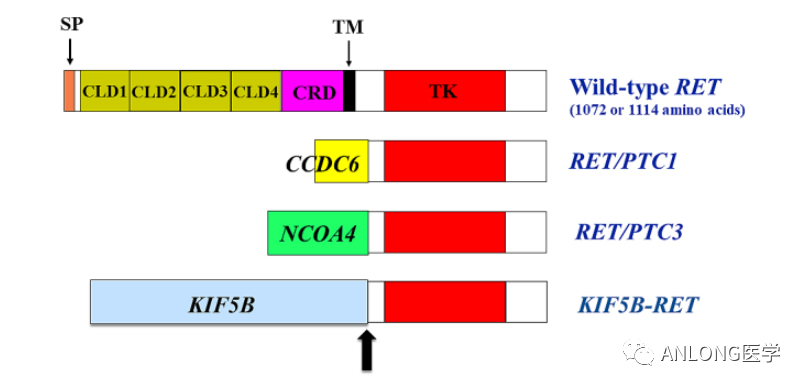
BRF117019和NCI-MATCH临床试验数据 X2101研究是一项多中心、开放标签、多队列试验,包括难治性或复发性实体肿瘤儿童患者。研究结果显示,达拉非尼联合曲美替尼的ORR为25%。对于9名有反应的患者,78%的患者DOR为6个月或更长时间,44%的患者DOR为24个月或更长时间。 RET RET原癌基因位于第10号染色体长臂(10q11.21),编码一种具有酪氨酸激酶受体RET蛋白。RET蛋白与配体结合后,可激活下游多种信号途径,如RAS、PI3K、STAT等,参与细胞的存活、增殖和分化。RET基因激活的RET蛋白通过多种信号通路参与不同肿瘤细胞的增殖、凋亡、侵袭,影响肿瘤的发生发展。 RET基因改变和野生型基因过度表达会驱动肿瘤形成,RET基因改变主要包括RET融合和点突变,其中肿瘤的形成主要与RET融合相关。目前已在多个癌种中发现RET融合,包括60%甲状腺髓样癌、10%~20%乳头状甲状腺癌、1%~3%非小细胞肺癌,在食管癌、结直肠癌、唾液腺癌、卵巢癌中的发生概率不足1%。在非小细胞肺癌中,RET基因融合阳性多见于年轻、女性、少量吸烟或不吸烟的亚裔患者。乳头状甲状腺癌中最常见的RET融合类型为CCDC6-RET和NCOA4-RET,而非小细胞肺癌中最常见的为KIF5B-RET。RET融合的检测方法有多种,如IHC,FISH,RT-PCR,DNA-NGS,RNA-NGS等,其中DNA-NGS和RNA-NGS检出率较高(RET基因融合可以用DNA-NGS检测到,但联合RNA-NGS检测可以提高检测灵敏度)。
RET基因主要融合类型 2022年9月21日,FDA加速批准塞普替尼(Selpercatinib,Retevmo)用于治疗携带RET基因融合的局部晚期或转移性实体瘤成人患者,这些患者在之前 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号