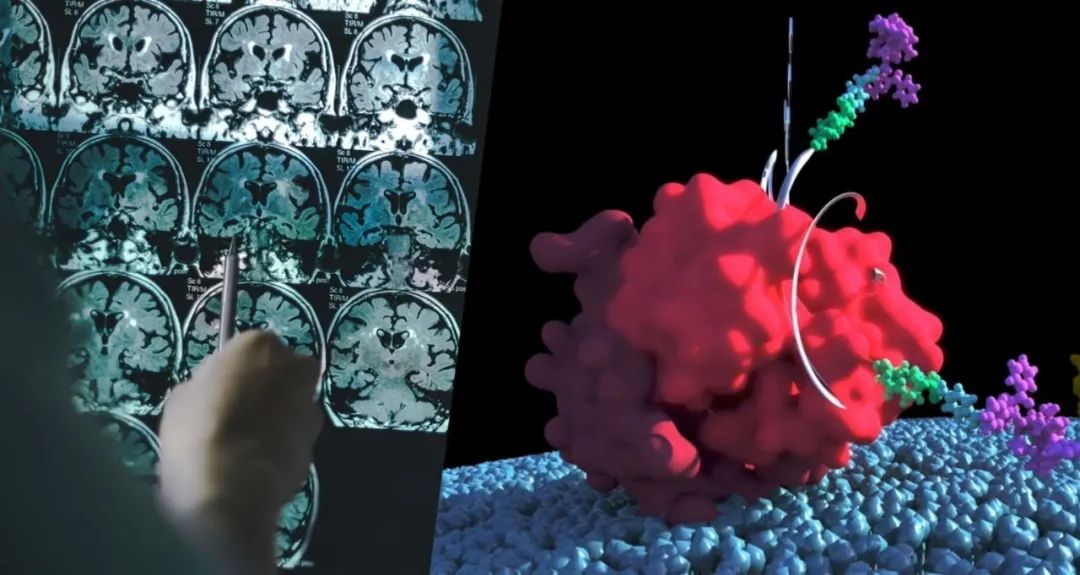
FDA于5月3日表示,它已经通过富士瑞必欧诊断公司(Fujirebio)的Lumipulse G β-淀粉样蛋白比率(1-42/1-40)测试的De Novo分类请求,允许该公司将其作为诊断阿尔茨海默病的工具进行销售。
该测试旨在用于55岁及以上有认知障碍、正在接受阿尔茨海默病和其他认知衰退原因评估的人,是FDA允许上市的第一个体外诊断工具,用于早期检测与阿尔茨海默病相关的淀粉样斑块。 传统上,临床医生使用PET成像技术检测这些斑块,每次扫描可能需要花费数千美元。Lumipulse测试通过测量患者脑脊液(CSF)中β-淀粉样蛋白1-42和β-淀粉样蛋白1-40的比率来检测淀粉样斑块的存在。 在为De Novo分类请求的临床研究中,研究人员查看了阿尔茨海默病神经影像倡议样本库中292份CSF样本的临床研究,并确定97%的斑块检测阳性者在PET检测中也是阳性,而84%的检测阴性者在PET扫描中也是淀粉样物质阴性。 FDA设备和放射健康中心主任Jeff Shuren在一份声明中说:“我们现在拥有了一种体外诊断测试,这有可能消除对耗时和昂贵的PET扫描的需要,对担心阿尔茨海默病诊断可能性的个人和家庭来说是个好消息。” “有了Lumipulse测试,就有了一种新的选择,测试通常在一天内完成,为医生提供关于大脑淀粉样蛋白状态的信息,但没有辐射风险,这将帮助确定患者的认知障碍是否是由阿尔茨海默氏病引起的。” Fujirebio在2017年获得了该测试的CE认证。据该公司称,在欧洲和其他接受监管指定的地区,每年有超过5万个与医院相关的记忆诊所正在进行这种测试。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号