高通量测序(以下简称NGS)技术是继Sanger测序后的一项革命性进步,能够同时对几十万到几百万条DNA进行测序,可实现在较低成本下对多基因、全外显子,甚至全基因组进行多种变异类型检测,且所需样本量和检测周期不会增加[1]。目前,NGS技术已广泛应用于肿瘤基因诊断、遗传病诊断和病原微生物诊断等领域。 由病原微生物引起的感染性疾病是全球死亡率和发病率较高的疾病之一。随着新冠病毒的爆发,如何快速准确检测病原微生物已成为传染性疾病早期诊断、精准用药和预后评估的首要问题。常规的院内检测主要依赖于微生物培养、形态学鉴定、生理生化特征分析及血清法免疫应答。这些方法均存在检测周期长、操作复杂、灵敏度及特异度受限等问题。而荧光定量PCR技术虽解决了上述问题,仍存在对未知病原微生物无法检出、通量低等问题。
近年来,随着测序成本的不断降低,NGS技术因其通量高、整体周期短等特点,可更好的应用于病原微生物分类及快速鉴定、传染病监测和耐药性研究。NGS 在病原微生物中检测主要采用两种方案,即rRNA基因测序和全基因组测序[2-3]。
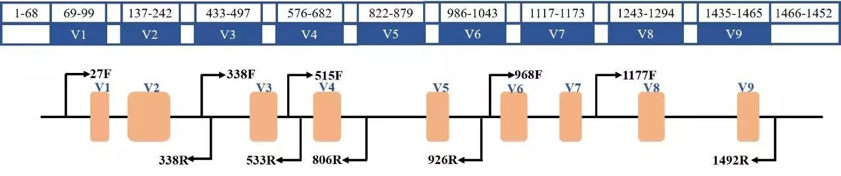
NGS应用于病原微生物的两种方案 针对宏基因组和16S 扩增子技术在病原微生物检测中应用的实例,小编将用2期文章带领大家深度探究。 rRNA基因测序(16s rRNA) 16S rRNA由于通量高、成本低、数据量少、分析简便等特点,更适用于研究样本中细菌的分类(属水平)及其在群落中的丰度变化。16S rRNA包括 10 个保守区域和 9 个高变区域,对高变区核酸序列进行NGS测序, 可区分属特性,用于细菌的分类鉴定和系统进化。16S扩增子测序通常是选择某个或某几个变异区域,利用保守区设计通用引物进行文库扩增,然后对高变区进行测序分析和菌种鉴定,16S rDNA扩增子测序技术已成为研究样品中微生物群落组成结构的重要手段[4-6]。
16S rDNA基因区域 实例一 George等(2017)利用半导体测序平台探究不同扩增子区域、不同数据库对16s rRNA技术在临床细菌多样性鉴定中的影响。该研究利用41种已知单菌检测4种16s不同高变区;结果表明,扩增片段最长的V1-V2高变区(313bp)可将这41种单菌细分鉴定到种,而其他三种单一区域的扩增方法大多仅能将细菌鉴定到属。因此,利用长读长(SE400、SE600)测序策略对16s V1-V2区进行NGS测序在临床上鉴定细菌是一种较好选择[7] 。
4种扩增子鉴定41种菌多样性鉴定结果 实例二 Laura等(2019)利用半导体测序Ion S5平台进行16s不同高变区单一测序来鉴定PNA系统中微生物群落信息。研究表明,各引物对靶向的高变区所获得的微生物群落组成的综合信息有显著差异(p < 0.001)。根据这项研究和以往的文献,针对微生物组鉴定没有单一的最佳引物。因此,作者建议使用多个16S rRNA 基因高变区引物对组合测序对微生物群落的鉴定具有重要意义[8]。
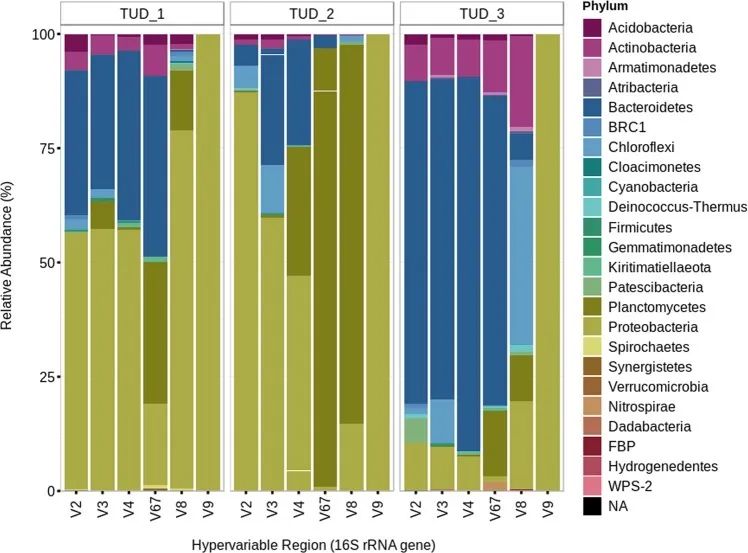
3个不同样本不同16s rRNA区物种多样性 实例三 Sema等(2019)利用半导体测序 Ion S5平台对20个吸烟者和非吸烟者的口腔拭子样本进行16s rRNA 7个可变区(V2、V3、V4、V6-7、V8和V9)panel 测序。结果表明,7个可变区联合分析提供的物种多样性最丰富;在单一区域中,V2区物种多样性较高[9]。
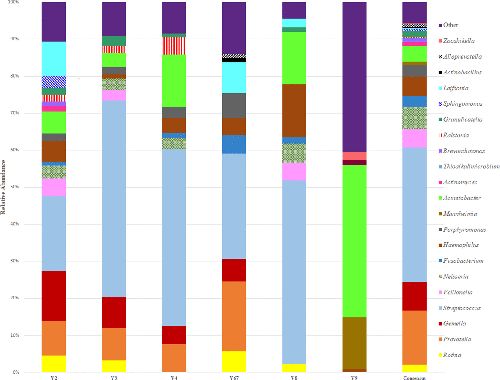
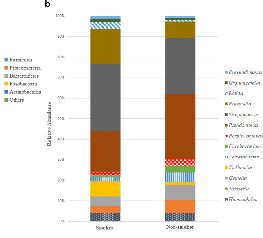
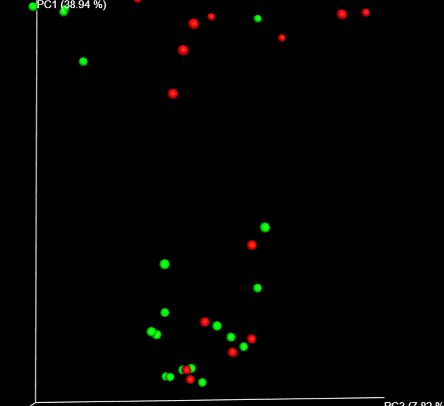
不同16s rRNA高变区物种多样性展示 与非吸烟者比较,放线菌和韦荣氏球菌在吸烟者中物种丰度增加,呈显著差异(P-adj=0.0390);Beta多样性组间差异分析的箱形图显示,吸烟者和非吸烟者存在组间互斥。因此,吸烟会改变口腔中微生物群落。
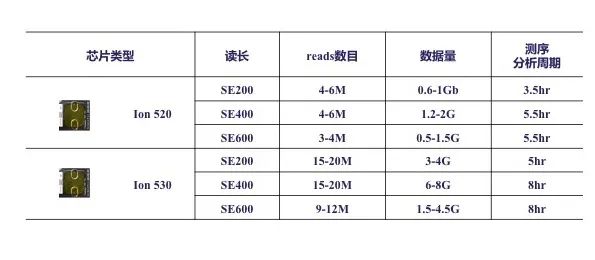
左图为吸烟者和非吸烟者微生物种群多样性 右图为Beta多样性吸烟/非吸烟组间差异图 GENETRON S5相比于其他测序平台,可实现单端400bp或600bp测序,无需overlap拼接,更适合于16S扩增子测序分析,为微生物多样性研究提供更多选择。
参考文献 [1] Erwin L. van D., et al. (2014). " Ten years of next-generation sequencing technology" .Trends in Genetics. 30 (9) :418-426 [2] Ruud H. D., et al. (2017). " Application of next generation sequencing in clinical microbiology and infection prevention " .Journal of Biotechnology. 243:16-24 [3] Stefan A. Boers., et al. (2019). " Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory " .Eur J Clin Microbiol Infect Dis .38 (6) :1059-1070 [4] Caporaso J. Gregory., et al. (2011) " Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample" .Proceedings of the National Academy of Sciences 108.Supplement 1 : 4516-4522. [5] Youssef Noha., et al. (2009) " Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys ". Applied and environmental microbiology. 75(16): 5227-5236. [6] Hess Matthias., et al. (2011). " Metagenomic discovery of biomass-degrading genes and genomes from cow rumen". Science 331: 463-467. [7] George S. Watts., et al. (2017). " 16S rRNA gene sequencing on a benchtop sequencer: accuracy for identification of clinically important bacteria". J Appl Microbiol 123(6): 1584-1596. [8] Laura orschler ., et al. (2019). " on resolving ambiguities in microbial community analysis of partial nitritation anammox reactors ". SCIENTIFIC REPORTS.9(1):6954. [9] Sema Karabudak., et al. (2019). " Analysis of the effect of smoking on the buccal microbiome using next-generation sequencing technology ". JOURNAL OF MEDICAL MICROBIOLOGY.68: 1148-1158. 声明:仅供专业人士参考 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号