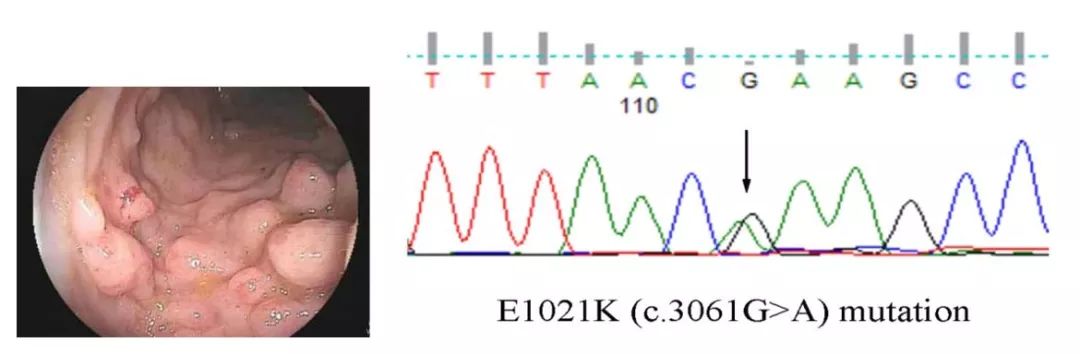
据称全球罕见病患者超过3亿。近年我国逐渐重视罕见病诊疗,发布了国家《第一批罕见病目录》和《罕见病诊疗指南(2019年版)》,一线医生、基因检测机构及药企和公益组织、爱心人士等积极参与。2019年2月28日是第11个国际罕见病日,数家专业机构和医生,从6个罕见病诊疗真实故事出发,抛砖引玉,发起“呐罕者计划”,邀请更多机构共同参与“罕见病诊疗可及性科普”中。欢迎留言探讨和报名。 1确诊前被怀疑是疫苗惹的祸 小A在8个月时出现发热、腹泻、口腔溃疡等症状,当地医生诊断为口腔炎,治疗后体温恢复正常。但是好景不长,2天后再次出现发热,伴皮疹、口腔溃疡、糜烂。爸妈怀疑是小A发病前接种的麻疹疫苗导至发病。小A辗转多家医院就诊,反复的出现发热、咳嗽、咳痰、腹泻,3岁后还出现肝脾肿大及贫血等。在5岁时来到复旦大学附属儿科医院免疫科就诊。免疫功能检查发现小AIgM显著增高,CD4+/CD8+T细胞比例倒置。B超发现肝脾明显肿大,胸部CT发现两肺下叶局部支气管扩张,胃肠镜检查结果发现结肠小肠多发隆起型病变。 医生高度怀疑是免疫缺陷病,基因检测结果发现小A存在PIK3CD基因突变,最终诊断为活化的PI3K综合征1型(APDS1)。小A接受了靶向药物西罗莫司治疗,1个月后感染及腹泻完全消失。目前治疗已1年多,整体状况恢复良好。
左图,胃肠镜检查结果发现结肠小肠多发隆起型病变;右图,基因检测结果 ◆ 病例来源:复旦儿科临床免疫中心,在中国大陆最早独立开展原发性免疫缺陷病基因诊断并应用于临床。建立了多种原发性免疫缺陷病的诊治规范(如XLA、CGD等);在中国最早成立了原发性免疫缺陷病慈善基金组织,并率先主导推动和实施了部分原发性免疫缺陷病治疗的医保报销制度;建立了卡介苗病的临床诊治流程,并阐述了其免疫相关机制;是我国食物过敏、预防接种不良反应、反复呼吸道感染的诊治等重要的先驱者之一;在国内率先对某些免疫缺陷病进行靶向治疗,是国内最大的原发性免疫缺陷病临床中心。 PI3Kδ过度活化综合征(APDS)有两种亚型:APDS1与PIK3CD基因突变有关;APDS2与PIK3R1基因突变有关,分别为常染色体显性和隐性遗传的免疫缺陷病。APDS1常见表现包括反复细菌或病毒所致反复呼吸道感染、支气管扩张、淋巴结节样增生、自身免疫等。(参考自中华儿科杂志, 2017,55(01): 19-24) 2十年辗转治疗,基因检测助力确诊 顾大夫在“运动障碍与神经遗传病”专病门诊曾经接诊一对姐弟,自幼患病,姐姐21岁,弟弟11岁,共济失调,智力较同龄人稍差,父母非近亲,无疾病表现。就医史十余年,两孩子的病情一直无法确诊,经干细胞治疗多次,还去过日本和台湾,已经花费500万元人民币。数月前在北京一家三甲医院神经内科就诊,送检全外显子测序,经过一番周折之后,找顾大夫解读基因检测报告。姐弟均携带MRE11A基因的一个已知致病突变(错义)和一个剪切突变(+2),分别来自父母。最终确诊为MRE11A基因突变引起共济失调伴毛细血管扩张样病1型(ATLD1)。就医多年,却一直没有做康复治疗,已严重足内翻、跟腱挛缩,久病未成医,基因检测让这个家系获得了明确诊断。 ◆ 病例来源:顾大夫,顾大夫工作室创始人,中文人类表型标准用语联盟(CHPO)总协调人,中日友好医院神经内科分子遗传实验室负责人,从事多年运动障碍与神经遗传病临床诊疗、科研、基因分析和患者教育公益工作。 遗传性共济失调(hereditary ataxias)占神经遗传病的10~15%,包含一大类遗传性神经退行性疾病,具有高度的遗传异质性和临床变异性,至少包括200多种疾病,文中案例即属其中一种,目前基于遗传模式和致病基因进行分型和诊断。从遗传模式上可分为:1、常染色体显性共济失调(ADCA),主要指脊髓小脑共济失调(SCA),还包括发作性共济失调(EA);2、常染色体隐性共济失调(ARCA),上文提到的共济失调伴毛细血管扩张样病1型属于这个类型,部分ARCA疾病以SCAR系列命名;3、X-连锁共济失调;4、线粒体母系遗传共济失调。(受访医生提供) 3被家人误解的“病秧子” 46岁的张女士因低血钾、乏力、关节痛、烦渴、夜尿增多、记忆力减退等症状来到吉林大学第一医院基因诊断中心就诊,这些症状已持续多年,在别人眼里,她是病秧子、娇气、懒惰,甚至家人也不理解,给她心理上带来很大的压力。来到吉林一院就诊后,医生为其进行临床全外显子检测,该检测项目覆盖人体十大系统与疾病密切相关的4450个基因的编码区,涵盖85%以上遗传疾病。检测结果显示,患者在SLC12A3基因上有两个致病性突变位点,分别来自其父母,确诊为Gitelman综合征。而她的妹妹们每人只携带一个突变位点,因此不会发病。 基因层面确诊使治疗有据可循,张女士进行门冬氨酸钾、氯化钾、醛固酮受体拮抗剂、消炎痛等联合治疗后,效果良好。确诊后也得到了家人的理解,极大地减轻了患者的身心压力。 ◆ 病例来源:吉林大学第一医院基因诊断中心,是目前是吉林省唯一一家具备资质能将高通量基因测序技术应用到临床的基因诊断中心,开展了包括肿瘤、遗传病、个体化用药生育健康类、病原微生物类5大类160余种基因检测项目,为临床快速诊断、精准治疗提供依据技术支持,填补了吉林省基因检测领域的空白。已获批吉林省精准医学示范中心、吉林省基因诊断工程实验室、吉林省基因诊断工程研究中心和基于基因检测技术的中药免疫效应重点研究室等。 Gitelman综合征(GS)是一种常染色体隐性遗传的失盐性肾小管疾病,临床特征为低钾代谢性碱中毒伴低镁血症和低尿钙症,患病率约为1-10/40000。常于青少年或成年发病。临床表型包括夜尿症、视力模糊、遗尿症、手足抽搐、肌无力、低钾性碱中毒、阵发性发热、烦渴、心悸、眩晕、关节疼痛、疲乏等多种表现。(参考自Gitelman综合征诊治专家共识.中华内科杂志, 2017,56(09): 712-716.) 4连续5次失败生育,竟是罕见病作祟 陆军军医大学第一附属医院妇产科产前诊断中心近期接诊了一名孕妇,连续5次不良孕史,其中3次孕晚期均出现胎儿NF增厚、侧脑室增宽、胸腹腔积液,最近一次妊娠,胎儿早产,并因颅内出血和颅脑结构畸形于生后几天便夭折,尽管产前超声和新生儿期的临床表现都疑似胎儿宫内感染,但孕妇和患儿却均未检测到感染证据。医院排除患儿核型与染色体微阵列芯片异常后,联合患儿及夫妇进行了家系全外显子测序分析,结果发现患儿存在Pseudo-TORCH综合征相关疑似病理性变异,由于患儿症状符合该综合征的临床表现,再获取前2次胎儿产前标本对变异进行验证,检测结果证实了上述推测。对于本案例中生育道路上历经磨难的这对夫妇而言,基因检测技术揭示了问题根源,合理的生育规划可以避免他们再次跌入同一条悲剧之河。 ◆ 病例来源:陆军军医大学第一附属医院妇产科产前诊断中心,是全军计划生育优生优育技术中心,重庆市首批成立的产前诊断中心。立足于军地服务,经过多年的发展本中心已建立完整的产前筛查、产前诊断、孕前筛查以及新生儿筛查技术平台,具备较为出色的遗传病诊断与咨询、产前筛查与诊断、胎儿宫内治疗经验和能力。 Pseudo-TORCH 综合征是临床表现类似于先天性TORCH综合征的一类多系统受累的遗传性疾病,其中2型临床特征包括胎儿宫内时期颅内出血、钙化、脑结构畸形、肝功能不全,常伴有血小板减少。患者往往有呼吸功能不足和癫痫发作,通常婴儿期死亡。该病的责任基因是USP18,遗传模式为常染色体隐性。(受访医生提供) 5年轻妈妈突发罕见病 2019年春节期间,发生在北京一位年轻妈妈身上的事情引起了业内关注。28岁的王怡涵戴着呼吸机蜷缩在病房一角,目前体重仅34公斤。3个月前,王怡涵还是个正常人,发病仅十来天就住进了ICU。丈夫为救妻千里迢迢从云南开车到北京大学人民医院神经内科就诊。10余天后基因检测得出结果:GAA基因复合杂合突变,辅助临床确诊为糖原累积症II型(庞贝病)。虽然得到了确诊,这个家庭却陷入深深的困境,一方面缺药,最新一批药物最快要等到3月份;另一方面药价昂贵,一年治疗费用约200万。等药期间,北京大学人民医院带头捐赠了一台呼吸机。大年初一是王怡涵孩子的2岁生日,但是这位年轻妈妈却还在苦苦等待天价救命药,命运未卜。 ◆ 病例来源:北京金准基因。 庞贝病为一种溶酶体贮积症,以常染色体隐性方式遗传,人群发病率仅四万分之一,根据发病时间可分成婴儿型和晚发型。由于位于第17号染色体上编码酸性α-葡糖苷酶的基因突变,造成体内酸性α-葡糖苷酶缺乏,糖原不能正常代谢而贮积在肌肉细胞的溶酶体中,导至严重的神经肌肉病变。目前有效的治疗方法为ERT(酶替代疗法),可采用赛诺菲健赞公司生产的药物美尔赞(单支售价5645元,需终身用药)。(参考自《中国当代儿科杂志》和北京金准基因) 6误诊十几年的“脑瘫患儿” 出生在一个普通家庭的两个患儿在被诊断为“脑瘫”的十几年后,一次偶然的机会,母亲将两个孩子的日常生活发布在某视频平台上,幸运的是被湖南妇幼保健院的遗传研究团队成员无意间看到,职业的敏感度让他们觉得,一个家庭有两个“脑瘫”的孩子,而且症状非常相似,“这可能不是简单的脑瘫,应该是由遗传因素导至的疾病”。在基因检测技术的帮助下,发现两姐妹携带有GCH1基因突变,被确诊为多巴反应性肌张力障碍(DRD),这是一种由于基因突变导至的代谢失调性的遗传性疾病,而非脑瘫。最终对症开出的药物每月仅需100多元,为这个家庭减轻了经济负担。服药1个月后可以自己吃饭,服药50天就能自己玩手机、开直播,两姐妹正逐渐走向康复。 ◆ 病例来源:贝瑞基因。 多巴反应性肌张力障碍(DRD)是以肌张力障碍或步态异常为首发症状的罕见遗传性疾病,发病率约0.5/100万,典型临床表现为儿童期起病,局灶性或节段性下肢肌张力障碍伴行走困难,可伴有轻度的帕金森氏综合征表现,如运动迟缓、强直等。与脑瘫患儿不同的是,DRD患儿一旦能够早期得以确诊并及时获得正确治疗,便能够有效地避免由多巴反应性肌张力障碍导至的肢体残疾。(参考自《医学综述》) 文章最后,特别感谢周文浩院长、姚宏主任、顾大夫、姜艳芳主任和马永毅博士等支持。 在接受采访时,医生们反馈国内罕见病诊治仍面临多种困境,如缺乏系统的医学科普平台、信息分散、缺乏有效整合、基层医生专业知识不足等。在2019年国际罕见病日的前一天,国家卫健委组织卫健委罕见病诊疗与保障专家委员会办公室(中国医学科学院北京协和医院)牵头制定并发布了《罕见病诊疗指南(2019年版)》。国家和社会各界爱心人士推动了我国罕见病诊疗的进步,未来罕见病诊疗规范化和科普之路仍任重道远。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号