压着2025年的尾巴(发布时间:2025-12-30),国家药监局器审中心(CMDE)发布了《体外诊断试剂变更注册审查指导原则》。一般来说,法规更新后,会尝试把最新发布的版本和历史版本进行一下比较,看看关注重点的变化。 目前在官网中查询,指导原则最早于2023-05-31发布了第一版的《体外诊断试剂变更注册审查指导原则(征求意见稿)》,当时法规适用范围为:本指导原则适用于体外诊断试剂的变更注册(触发程序上的变更),即医疗器械注册证及产品技术要求、产品说明书载明的内容发生变化的情况(触发条件)。本指导原则涵盖的变更情况包括体外诊断试剂变更注册申报资料要求及说明中综述资料部分产品变更情况描述的12种情况(触发条件)。 之后于2025年初(2025-01-24),进行了第二次公开征求意见,适用范围进行了一定程度的细化,变为:本指导原则适用于体外诊断试剂的变更注册,即医疗器械注册证载明的产品名称、包装规格、主要组成成分、预期用途、产品技术要求、产品说明书、进口体外诊断试剂的生产地址等发生变化的情况。本指导原则涵盖的变更情况包括《体外诊断试剂注册申报资料要求和批准证明文件格式》(2021年第122号)附件6(较上次明确)中综述资料部分产品变更情况描述的12种情况。 最终,2025年底发布的正式版指导原则,适用范围中,对于参考的法规又有了细化:本指导原则涵盖的变更情况包括《关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告》(2021年第122号)附件6中变更注册申报资料要求及说明下(较上次新增)综述资料部分产品变更情况描述的12种情况。 综上,“《关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告》(2021年第122号)附件6中变更注册申报资料要求及说明下综述资料部分产品变更情况描述的12种情况”也是需要我们一起关注的重点,并且是指导原则的一个基础。 后续我们在工作中,如果遇到需要评估是否需要进行变更注册的情形时,最先想到的,就应该是这12种情况。 根据产品具体变更情况提供相应的说明及对比表,包括下列情形:
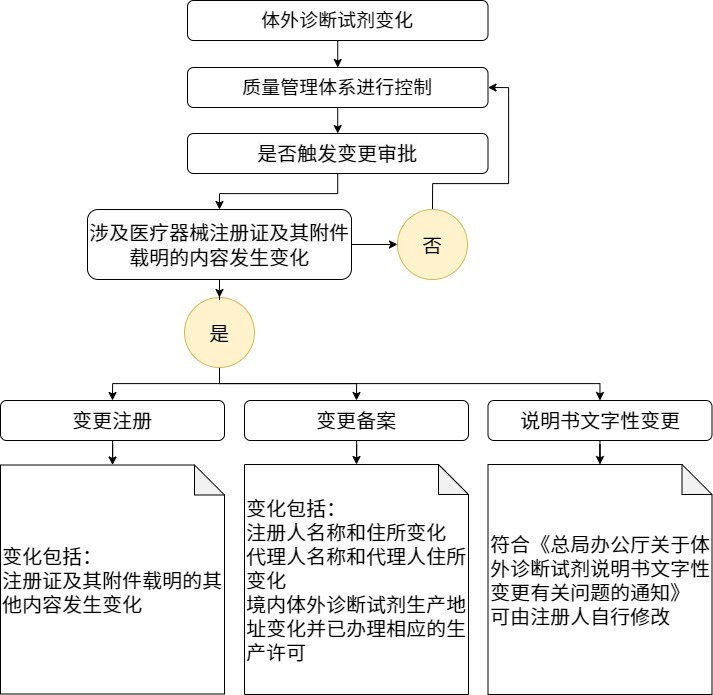
结合上述“12种情况”和终版的指导原则,我们来把相关的重点内容梳理一下。 指导原则包括7个大的模块,分别是: 01 适用范围 重点: 本指导原则适用于“体外诊断试剂的变更注册”,这里需要注意的有两点,一是说明本指导原则的范围,是触发了“程序上的变更”,需要提交相关资料给审评老师进行审批,二是不包含“变更备案”的情形。 法规明确指出变更注册的触发条件: ①医疗器械注册证载明的产品名称、包装规格、主要组成成分、预期用途、产品技术要求、产品说明书、进口体外诊断试剂的生产地址等发生变化的情况。 ②《关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告》(2021年第122号)附件6中变更注册申报资料要求及说明下综述资料部分产品变更情况描述的12种情况。 关于变更注册和变更备案的触发条件分别具体的内容,法规原文中在“3、体外诊断试剂变更管理流程”进行了描述。 针对提交评审资料中,涉及需要评估非临床与临床具体内容,是需要根据不同的变更,进行合理的风险分析评估后进行的。 02 体外诊断试剂变更管理的原则 重点: 生产质量管理体系持续有效运行和风险管理 这是进行变更管理最重要的两个基础,产品变更出现后,需要一个有效运行的体系来进行评估,并立即启动变更对于产品收益风险的评估,通过实施风险控制措施、受益-风险分析等手段进行风险控制,在结果显示变更引发的综合剩余风险可接受的情况下进行产品变更,并形成与产品变化相关的产品风险管理资料。 03 体外诊断试剂变更管理流程 重点: 如涉及医疗器械注册证及其附件载明的内容发生变化,需进行变更备案或变更注册;如不涉及医疗器械注册证及其附件载明的内容发生变化,注册人也应当按照质量管理体系要求做好相关工作。 04 体外诊断试剂变更注册审查要点 重点: 变更注册事项的情形较多,注册人需根据具体变更内容进行风险分析和技术评价,综合受益-风险分析,确定产品是否适宜进行变更,基于变更对产品安全性、有效性的影响,采用适当的方法进行相应的评价或研究并提交资料。 变更注册申报资料应参考第122号附件6中关于体外诊断试剂变更注册申报资料要求及说明。 指导原则内容为对于(一)综述资料、(二)非临床资料、(三)临床评价资料、(四)产品说明书、(五)质量管理体系文件等资料的详细要求。 另外,针对每个模块不同的侧重点,比如效期变化,需要重点关注非临床资料的稳定性研究,比如阳性判断值变化,可能会有触发重新进行临床评价的情形,具体需要根据产品变化来判定,需要提交哪些模块资料以及不同模块资料的重点研究方向。 这也就是下面法规重点需要关注的第五部分的内容。 05 体外诊断试剂的常见变更情形 重点: 和2021年第122号,附件6中变更注册申报资料要求及说明下综述资料部分产品变更情况描述的12种情况进行结合,进行详细的说明。 针对各种产品变更情形,分别描述对于单一变更情形进行分析、评价或研究的考虑因素以及涉及的部分重点申报资料的注意事项。 对于指导原则中,针对常见变更情形的考量以及提交资料内容这个模块是需要关注的重点,这部分的内容比较多,个人认为是比较详细的,每一种变更情形都会有对应的评价思路和申报资料的注意事项,大家如果需要可以自行对照法规的不同情形进行评估。指导原则中的很多内容是针对122号文中12种情况的细化,我们也可以参考法规,评估出在实际工作中和法规中举例的类似变更情形下的变更审评关注点。 以下仅为目前在我工作过程中,遇到过的一些常见的情形,在原文中抽取出来展示,详细内容大家可以参考法规原文。 06 不适用于体外诊断试剂变更注册的情形 重点: 对于核心反应体系原材料的抗原、抗体实质性改变(例如蛋白结构和序列、型别、生物学来源、单克隆或多克隆、克隆株、免疫刺激原等)或待测物的引物、探针核酸序列改变、增加核心反应成分、产品检验原理发生实质性改变等构成新产品的情形,应按照产品注册进行办理。 07 参考文献 指导原则相较于之前112号文的要求,进行了细化,尤其是针对每个变化情形,都会有对应的评价思路和申报资料注意事项,尤其是评价思路,可以学习并且延伸到很多地方,之前参加相关法规培训的时候,问答环节,针对变更注册新增适用机型、扩增预期用途范围、增加样本类型等等问题还是很多的,而且变化的情景确实多种多样,所以可能实际中你我碰见的,就是非常罕见的产品变化情景,超出了法规中明确说明的场景,这个时候,我们也许可以先吸收一下评价思路中的思维模式,把变更情况说明清楚,然后在有效的体系下,进行充分的风险分析,把应做的非临床与临床研究进行完善,最终呈现在我们的变更申报资料中,这也许才是对产品变更交出的一份满意的答卷。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号