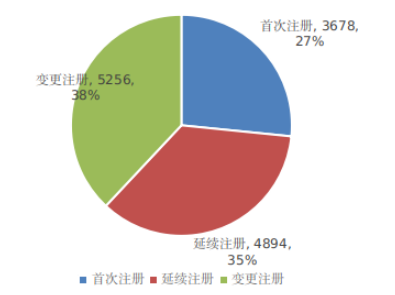
一、前言体外诊断试剂上市后,常常会遇到各种变更需求。这些需求可能源于市场反馈,如需要延长产品稳定性;或源于供应链变化,需要变更原材料;或为了改进生产工艺,优化产品质量;或为满足法规及标准要求,对检验方法进行变更。 根据《2024年度医疗器械注册工作报告》数据显示,变更注册约占注册申请的38%,首次注册申请占27%,延续注册申请占35%。相比首次注册和延续注册,变更注册可参考的法规文件较少,变更种类繁多,导至企业和审评需要投入更多精力。
国家局器械审评中心在2023年5月30日发布了《体外诊断试剂变更注册审查指导原则(征求意见稿)》,并在2025年1月24日发布了第二次征求意见稿。虽然尚未形成终稿,但2025年征求意见稿的内容已具有较高的可执行性。 二、变更的基本要求1.质量管理体系控制体外诊断试剂发生变化均应通过质量管理体系进行控制。即是建立变更程序→按文件执行→并保留相关记录。
2.变更备案或变更注册当涉及医疗器械注册证及其附件载明的内容发生变化,需向原发证单位提交变更备案或变更注册。
3.申报资料要求变更注册申报资料应参考《体外诊断试剂注册申报资料要求和批准证明文件格式》(2021年第122号)附件6中关于体外诊断试剂变更备案/变更注册申报资料要求及说明。 三、变更的不同情形1.低风险变更情形低风险变更情形是指不影响产品安全性、有效性的变化,申报资料要求相对简单。这类变更主要包括:
对于低风险变更,申报资料要求包括概述和产品变更情况描述,说明变更的原因并明确具体变更事项,必要时提交支持变更的相关资料。涉及产品技术要求或说明书变化的应列表说明具体内容。 2.有潜在风险变更情形有潜在风险变更情形是指可能影响产品安全性、有效性的变化,申报资料要求相对复杂。这类变更主要包括:
资料要求包括概述和产品变更情况描述、产品风险管理资料、临床前评价资料和临床评价资料。 根据变更可能影响的性能方面,需要提供相应的评估资料,如分析性能研究、稳定性研究、样本稳定性研究、样本适用性研究、生产工艺研究资料、反应体系研究资料或阳性判断值/参考区间研究资料等。 在以下情形通常需要提供临床评价资料:
3.不适用于变更注册的情形对于核心反应体系原材料的抗原、抗体实质性改变(例如蛋白结构、供应商变化)或引物、探针核酸序列改变、增加核心反应成分、产品检验原理发生实质性改变等构成新产品的情形,应按照产品注册进行办理。 五、结语更投入的时间和资金成本约为首次注册的一半,因此变更决策需要审慎。企业应通过质量体系中的变更程序控制申请、评估、验证、生效、实施等环节,确保变更决策合理。合理的变更管理不仅能够提高产品质量和性能,还能降低变更成本,提高企业的市场竞争力。 《体外诊断试剂变更注册审查指导原则》的发布,为企业提供了明确的变更注册指导,有助于企业更好地理解变更注册的要求和流程,提高变更注册的效率和成功率。企业应密切关注该指导原则的最终版本,并根据指导原则的要求,合理规划和实施产品变更。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号