1.印度经济前言
中国面积约960万平方公里 中国人口约14.43亿 中国人均GDP 12720.22 $(2022年) 印度2022年人均GDP与中国2006年人均GDP(2099.21 $)接近,所以说印度的经济发展相当于20年前的中国。正所谓“已有的事,后必再有;已行的事,后必再行。日光之下,并无新事”。中国20年前发生的,正在印度发生,高端设备依赖进口、国家鼓励和政策支持有一定技术门槛的医疗器械国产化,低技术低成本医疗器械竞争白热化。正如20年前的中国,医疗器械主要靠进口,印度近80%的医疗器械依赖进口,特别是高端设备,如癌症诊断、医学影像、超声波扫描和PCR设备,在印度,美国占市场份额15%。新加坡,德国和中国分别占7%,6.7%和6.4%,此外,美国,德国,中国,日本和新加坡是向印度出口高科技医疗设备的五个最大出口国。印度的IVD行业正迅速发展,印度IVD(试剂及配套设备)市场规模约15亿美元,并以每年20%的复合增长率增长。相比122亿美元的医疗器械市场规模,IVD占比12%左右。印度有750-800家国内医疗设备制造商,但大多数是中小企业,大约65%的制造商主要是国内企业,从事耗材业务,并满足出口受限的本地消费。2.印度医疗器械监管法规框架印度的医疗器械监管机构是Central
Drug standard control organization (CDSCO),相当于美国FDA,中国NMPA。
CDSCO下设中央许可证颁发机构Central Licensing Authority(CLA)和州许可证颁发机构State Licensing
Authority(SLA),CLA相当于国家药品监督管理局,SLA相当于各省药品监督管理局。CLA负责进口A、B、C、D类和国产C、D医疗器械的批准; SLA负责国产A、B类医疗器械的批准。 印度早期是没有专门的法规对医疗器械进行监管,1989至1995年期间少数医疗器械作为药品进行管理(避孕用品、皮下注射器、针头、输液装置、HIV体外诊断试剂、手术缝合线、药用胶带、外科辅料),多数器械无监管。 2005年10月6日制定《医疗器械市场进口与生产指南》,2006年3月1日正式实施,增加十大类器械进口或销售前需要经过CDSCO的许可(心脏支架、含药支架、导管、角膜镜、注射器、骨粘合剂、心脏瓣膜、静脉输液针、骨科植入物、人工假体)。 2017年1月,CDSCO发布了《India Medical Device Rules 2017》简称MDR(不是指欧盟Medical Device Regulation),于2018年1月1日正式实施,标志着进入印度医疗器械进入监管时代。 2020年2月,CDSCO对Medical Device Rules 2017进行了修订,发布了MDR Medical Device
(Amendment) Rules, 2020,于2020年4月生效。增加了新的条款规定某些需要注册的医疗器械要求。与全球法规协调一致,印度医疗器械也基于风险分类进行监管(A、B、C、D类),MDR中规定了分类基本原则,IVD产品的主要分为B、C、D类。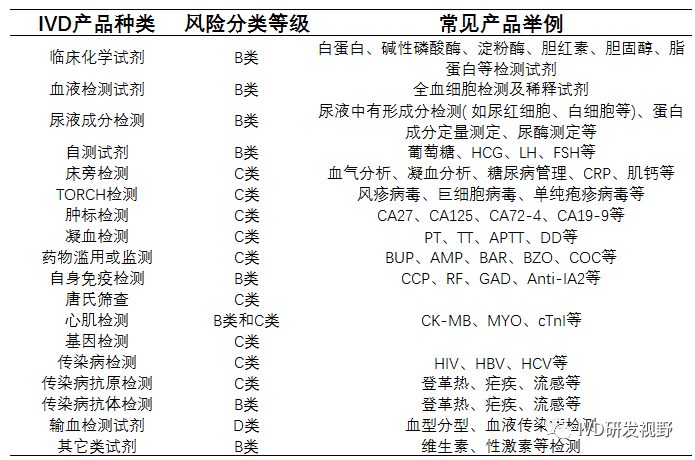
准确分类通过CLA发布的清单进行确认《Clarification On Import And Manufacture Of Medical Devices》,医疗器械注册提交文件格式要求参考《Guidance Document on
Common Submission Format for Registration of Medical Devices in India》。3.医疗器械许可申请费用印度医疗器械许可证申请收费取决于产品分类,如果产品属于创新/新型医疗器械,即在印度市场无同类产品(预期用途、技术特征类似产品)时,需要额外的费用及更长的时间,因为需要增加创新医疗器械进口/制造审批。不论产品种类,进口许可的时间周期在6-12个月左右。进口许可证永久有效,除非自动注销或是特殊原因取消。虽然永久有效,但需要每5年到期时缴纳许可证保留费用,保留费用同许可证注册费用一致。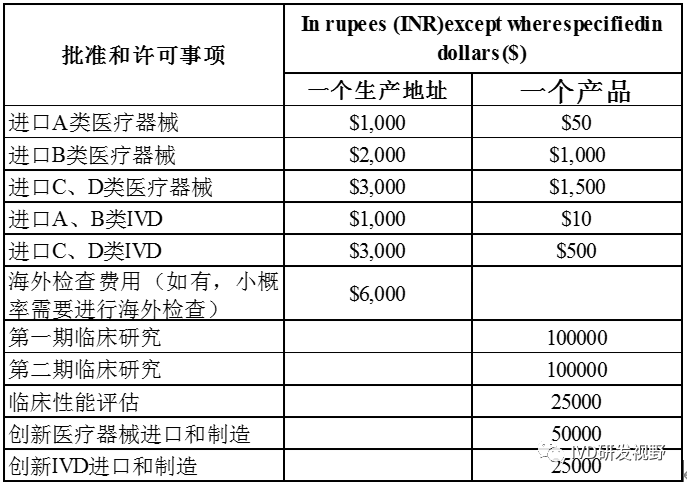
4.医疗器械申请许可需要文件申请医疗器械进口或制造许可证,需要提供以下文件,A类无需要提供产品主文件,体外诊断试剂需要提供性能测试报告,对于医疗器械进口还需要提供自由销售证书及其它资质证照。下述文件中的工厂主文件和产品主文件内容较多。
6.生产信息(流程图、作业指导书、灭菌过程、返工处理等)1.执行摘要 2.IVD描述、规格、及附件 3.安全性基本原则检查清单 4.风险分析与控制总结 5.设计和制造信息 6.产品验证和确认 7.分析性能研究 8.样本类型 9.分析性能特征。 10.分析灵敏度 11.分析特异性 12.计量溯源性 13.测量范围 14.临界值的建立 15.稳定性 16.产品有效期 17.使用稳定性 18.运输稳定性 19.临床证据 20.标签 21.上市后监管 22.需要提交的其他信息 4.3检验报告和临床试验进口医疗器械和IVD如果分类属于C类和D类时,需要在印度进行临床试验,如果器械已获得参考国(美国、日本、加拿大、澳大利亚、欧盟)的自由销售证书,可以免于临床试验。D类IVD需要在印度医疗器械中央测试实验室进行性能测试,B类和C类在认可的印度本国实验室进行性能测试,如果器械已获得参考国(美国、日本、加拿大、澳大利亚、欧盟)的自由销售证书,可以接受原有的检验报告。5.结语比较印度的IVD国内制造许可证和IVD进口许可证的获批流程和要求,可以看出有一定程度的本国保护,对进口测试报告和临床试验的要求严于国内制造,申请创新/新型C类和D类国内制造许可证时,才需要临床试验。
THE END
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 









