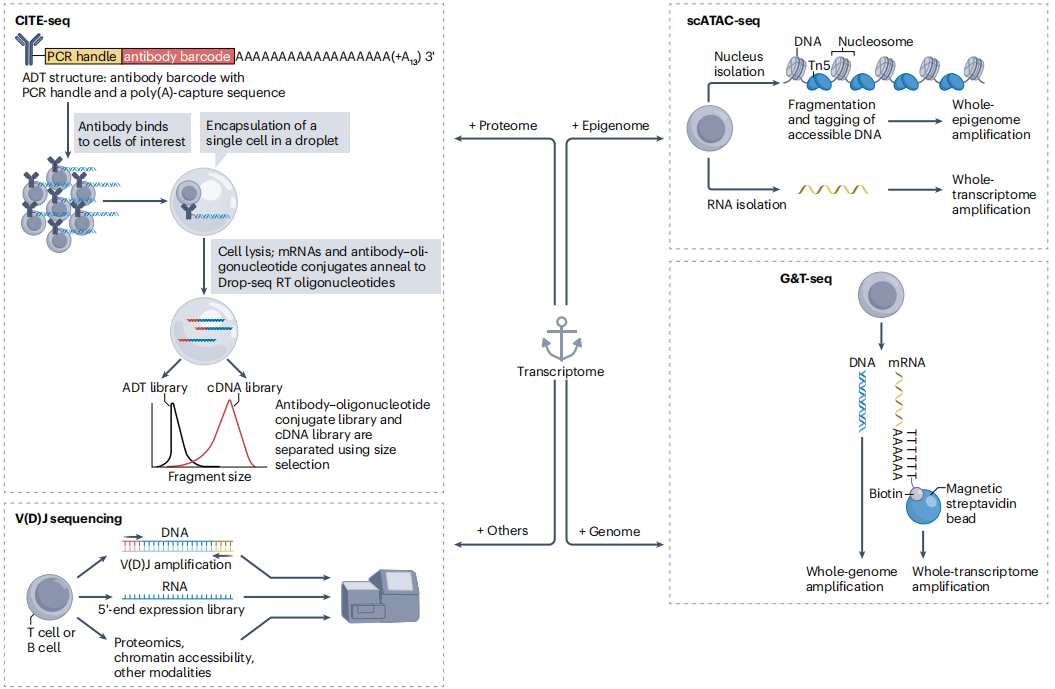
近日,一组来自耶鲁、纽约大学、纽约基因组中心的研究团队在杂志Nature Reviews Molecular Cell Biology上发表了一篇题为“The technological landscape and applications of single-cell multi-omics”的综述文章。在这篇全面的综述中,作者讨论了已建立的多组学技术以及该领域的前沿和最先进的方法,也讨论了多组学的局限性。作者强调单细胞多组学技术在细胞谱系追踪、组织特异性和细胞特异性图谱制作、肿瘤免疫学和癌症遗传学以及基础和转化研究中的细胞空间信息制图方面的影响。最后还讨论了已经开发的生物信息学工具,通过使用更好的数学建模和计算方法来连接不同的组学模式和阐明功能。 图片来源:Nature reviews molecular cell biology 一 单细胞多组学概述 各种单组学方法产生了大量的单模态数据,旨在剖析基因调控的机制基础或揭示人类疾病的各个方面。方法和技术的进步现在允许同时分析基因组、表观基因组、转录组、蛋白质组和其他新兴的组学模式,以更好地理解生物学机制和基因型与表型的关系。在过去的十年中,已经能够以单细胞的分辨率对细胞类型特异性基因组信息进行基因组尺度的表征。随着单细胞技术的不断进步,能够在组织内空间定位基因表达,即“空间多组学”。 整合基因组和转录组 最成熟的单细胞组学方法是单细胞转录组学,它通常与其他组学相结合研究基因表达与表型异质性之间的联系(如下图)。在单细胞全基因组mRNA测序(scRNA-seq)的基础上,多组学技术同时通过细胞多路复用、细胞标记、cell hashing和其他方法进行多模态复用并增加通量。 对同一单个细胞的基因组和转录组进行平行分析可以反映基因组的转录状态,提供关键突变的覆盖范围,提供细胞中的基因组-转录组相关性,这些相关性最终导至在疾病中发现新的基因表达程序。从2014年首次报道了利用微流体技术对单个细胞的DNA和RNA进行物理分离并进行全基因组和全转录组进行扩增和测序,到2019年,TARGET-seq方法显示,通过使用条形码,每次运行约5,000个单细胞的通量,并且显著提高突变覆盖率。 整合转录组和表观基因组 单细胞表观基因组分析与转录组学相结合,可以直接阐明DNA表观遗传特征,如DNA甲基化、DNA可及性和与它们产生的转录组相关的组蛋白修饰。单细胞三组学测序(scTrio-seq),可对基因组、甲基化组和转录组进行联合分析。scTrio-seq2结合scTrio-seq和scBS-seq来分析甲基化与mRNA和拷贝数变化的关系; scNOMe -seq表征核小体定位、染色质可及性和DNA甲基化。还有单细胞核小体占位图谱scNMT-seq和scNOMeRe -seq,可以分析DNA甲基化、染色质可及性和来自同一DNA分子的转录本。单细胞分辨率组蛋白修饰有助于理解细胞的表观遗传程序和分化轨迹,如scCUT&TAG-pro,用于分析蛋白质- DNA相互作用和单细胞中表面蛋白的丰度。这些方法已经扩展到使用多组学技术(如Paired-Tag和CoTECH)来表征RNA和组蛋白修饰,这些技术使用组合条形码来实现转录组和染色质占用的高通量检测。 多组学技术继续发展,以增加通量和多模态。中通量方法分析表观基因组和转录组,如scCAT-seq和ASTAR-seq。Sci-CAR-seq和SNARE-seq可检测超过11,200个细胞核的染色质状态和基因表达。TEA-seq,一种用于三模态单细胞转录本、表位和染色质可及性测量的方法,使用scATAC-seq工作流程和10x Genomics Multiome ATAC Plus基因表达试剂盒来阐明数千个单细胞中基因调控的调节因子。 整合转录组和蛋白质组 2017年,随着CITE-seq技术的发展,转录组和蛋白质组共分析的通量实现了巨大飞跃。该方法结合了高度复用的蛋白质标记检测的转录组分析,可用于数千个单细胞。其他不依赖抗体的单细胞蛋白质组学方法可以产生更全面的细胞蛋白质组特征,如质谱方法SCoPE-MS和SCoPE2,但它们还没有与其他组学集成的方法。PHAGE-ATAC是最近开发的一种基于纳米体显示噬菌体表位识别的方法。编码纳米体的吞噬体的高可变互补决定区3 (CDR3)作为一个独特的遗传条形码,通过下游测序识别,并作为抗原检测的代理。这种多组学方法可以对数千个单细胞进行分析,同时可靠地检测细胞表面蛋白、线粒体DNA基因型和表观基因组修饰。 RNA和免疫检测(RAID)是一种用于细胞内蛋白或磷酸化蛋白的免疫检测以及mRNA分析的方法,可以对环境刺激与异质细胞反应进行相关性分析。更多模态的是NEAT-seq,可同时描绘单个细胞中核蛋白表位、染色质可及性和转录组的丰度。最后,CRISPR兼容的CITE-seq (ECCITE-seq)扩展了蛋白质多组学领域,纳入了CRISPR兼容的转录组、免疫repertoire和蛋白质组索引,包括克隆型分析和CRISPR定向扰动。因此,多组学技术有能力测量细胞扰动和信号通路对单细胞状态的影响。 其他多组学模式 多组学的其他方式包括表征T细胞受体(TCRs)和B细胞受体(BCRs)的复杂基因组库,可以提供对个体免疫谱的深入了解,并提示各种受体结构如何促进适应性免疫反应、自身免疫和肿瘤生长。将特定的TCR和BCR (DNA)克隆型与scRNA-seq结合,为更好地理解抗原受体的转录组、克隆组成和独特序列特征(如CDR3长度和特定序列基序的存在)之间的关系铺平了道路。大多数能够在保持单细胞全转录组信息的同时捕获TCR和BCR异构体序列的技术都实现了5′端RNA扩增,该技术已由10x Genomics商业化,并已扩展到包括蛋白质组学(ECCITE-seq)和染色质可及性(T-ATAC-seq)(如下图)。免疫库测序,加上技术进步和与其他组学模式的整合,有能力更深入地了解T细胞和B细胞的克隆性和特异性如何影响免疫反应。
多组学测序的景观。 图片来源:Nature reviews molecular cell biology 02 多组学走向空间 新兴的空间多组学技术试图在基因组尺度上扩展完整组织内单细胞之间相互作用的图谱(下图)。解决这一前沿领域的重要性至关重要。 成像技术 荧光原位杂交(FISH)是一种自20世纪80年代以来就存在的技术。MERFISH通过组合标记和多重读出杂交的顺序成像,大大增加了单细胞中可以同时成像的RNA分子数量。seqFISH+展示了对组织空间组织进行发现驱动研究的能力,它能够成像单个细胞中10,000个基因的mRNA,可获得转录组水平的读数。为了评估RNA和蛋白质,RNAscope将DNA与金属标记的探针杂交,然后将金属结合抗体添加到组织切片中,并用质谱流式细胞技术测量金属丰度。 空间蛋白质组学领域也随着CODEX等技术的发展而扩大,CODEX使用多路细胞成像方法定量表征蛋白质表达并定义组织结构。最近,一种自动化的集成平台——空间分子成像仪被开发出来,用于在单细胞和亚细胞分辨率下解析组织中980种RNA和108种蛋白质的多组学空间分布,对荧光分子条形码进行多周期的核酸杂交。 Sequencing-based技术 基于测序的技术可以直接在完整组织内感兴趣的生物分子(DNA, RNA或蛋白质)上执行。2016年,空间转录组学被引入,通过从组织中提取分子,从原位测序转向非原位测序(下图b)。在完整组织上进行反转录,得到的cDNA在组织裂解前与载玻片上的100 μm阵列寡核苷酸偶联,最终生成NGS文库。后来,微米级的bead阵列被开发出来,用于在细胞水平上捕获空间转录组学数据(下图a)。GeoMx(来自Nanostring)使用一种靶向FISH检测方法,通过光切割和DNA探针测序,可以在组织的一个选择的微观区域检测多达10,000个基因,这些DNA探针与目标基因杂交,探针面板现在接近全转录组覆盖水平。 2020年报告了一种完全不同的空间组学测序方法。DBiT-seq微流控方法允许以空间分辨的方式将DNA条形码传递到组织切片中,并创建10-50 μm空间条形码像素(下图a)。DBiT-seq是第一个报道的空间多组学技术,显示了数十种蛋白质和整个转录组的共定位,已进一步扩展到染色质可及性和组蛋白修饰的空间表观基因组分析。 激光捕获显微解剖技术 LCM用于分离微米级组织甚至单细胞,同时保留空间信息从而将组织学与分子测量联系起来。一项通过mRNAs、microRNAs、DNA甲基化和蛋白质表达共同分析构建动态调控网络的工作证明,基于LCM可以同时表征多种模式。如Geo-seq所示,LCM还可以与其他技术结合来阐明细胞异质性和空间变异。最近,LCM与荧光成像结合使用质谱法对福尔马林固定石蜡包埋的组织进行了深入的蛋白质组学表征。基于LCM的方法允许在单细胞水平上进行完整的基因或蛋白质分析,包括位置信息,但它们只能检测少量细胞。尽管在单细胞水平上存在通量限制和无法逐像素绘制整个组织切片,但LCM具有多功能性,可以分离空间定义的组织区域进行多组学分析。
走向空间分辨的多组学。 图片来源:Nature reviews molecular cell biology 03 单细胞多组学的研究与临床应用 细胞谱系追踪 单细胞测序的一个主要应用是建立和构建疾病进化的细胞谱系树或系统发育。最近的高通量、多路测序技术为谱系追踪铺平了新的方向。上述方法,如scTRIO-seq和scNMT-seq,可用于同时对单个细胞的基因组拷贝数变化、DNA甲基化组、核小体占用和转录组进行采样,以揭示新的细胞类型及其在研究谱系中的作用。单细胞蛋白质组学方法可用来分析谱系特异性转录因子及其随时间的丰度。最近开发的iTracer在诱导多能干细胞衍生的类器官中结合了报告基因条形码和可诱导的CRISPR-Cas9,并且与单细胞和空间转录组学兼容,可用于克隆追踪和不同时间点的谱系记录。 最后,结合基因组、转录组和谱系报告方法,可以在癌症等疾病中进行谱系追踪。追踪癌症细胞谱系,可发现哪一部分谱系优先受到药物治疗的影响,以及参与的细胞信号通路;还可以了解免疫细胞如何对感染作出反应,如何分化以及命运背后的机制。最近的一项研究表明,结合scRNA-seq和scTCR-seq来追踪CD8+ T细胞的克隆扩增和分化,最终描绘导至细胞耗竭的途径。谱系追踪也可以结合scRNA-seq和其他单细胞模式进行空间分辨率,以表征细胞组织动力学并区分分子特性和形态。总之,单细胞多组学对疾病的细胞谱系分类、肿瘤分类以及我们对细胞状态和类型进化的理解产生了重大影响。 器官的细胞类型图谱 癌症基因组图谱(TCGA)是一个癌症特异性多组学数据资源,表征了来自33种癌症类型的20,000多种原发性癌症和相应的正常样本,代表了基因组、表观基因组、转录组和蛋白质组学模式的数据。DNA元件百科全书(ENCODE)项目联盟于2003年开始试点,此后扩大规模,使用多组学测序来定义和注释人类和小鼠基因组中的功能元件。还有研究旨在提供心脏、肺、血液、睡眠障碍和衰老的综合多组学图谱,所有这些图谱都包含数千个数据集,以确定与疾病发病和进展相关的细胞类型。 第一个通用的人类细胞图谱项目于2017年启动,旨在提供一个开放获取的资源,用于将所有单细胞组学数据整合到一个图谱中,以加深对细胞发育、生理和细胞间相互作用的理解,并预测细胞扰动或突变对人体每种细胞类型的影响。项目数据收集主要采用单细胞基因组学方法,包括单细胞多组学和空间技术。近年来,空间多组学数据被用于通过整合多种互补技术为细胞基因组注释添加组织背景。这种组合为图谱的生成增加了一个有价值的参数,因为细胞的空间分布可以将特定细胞类型的多组学特征与其在组织中的定位联系起来。 肿瘤免疫学和癌症遗传学 单细胞多组学技术在免疫肿瘤学研究中产生了更全面和变革性的见解,包括但不限于定义肿瘤和免疫细胞状态,揭示它们在特定疾病背景下的相互作用,推断抗原-免疫受体动力学的复杂性,确定治疗反应的预测性生物标志物,并为多种癌症类型的治疗方法的发展提供方向。 一项新开发的空间技术slide-DNA-seq被应用于小鼠转移模型和原发性人类癌症,揭示了空间上不同的克隆群体,揭示了与克隆变异和局部肿瘤微环境相关的基因。使用多组学方法也研究了癌症进展的机制。通过对信号转导因子SMAD4失活对转录组、蛋白质组和分泌组的影响的研究,发现了可能促进晚期结直肠癌个体转移的三个SMAD4介导的过程。另一项研究将DNA可及性、基因表达和蛋白质丰度的单细胞多组学整合应用于混合表型急性白血病模型,确定编码转录因子的RUNX1基因是与低生存率相关的混合表型急性白血病的潜在致癌基因。 这些方法也被用于研究免疫检查点阻断(ICB)治疗人类癌症。一项研究将Akoya Biosciences的空间表型和免疫效应因子面板与多重免疫荧光成像、scRNA-seq和TCR库分析结合起来,阐明了三阴性乳腺癌中响应ICB的T细胞逃逸途径。作者预计空间和单细胞多组学技术的更广泛应用以及计算方法将创建一个综合框架,用于理解癌症异质性的复杂本质。 04 用于多组学数据集成的计算工具 该领域的一个主要障碍是多组学数据集成的固有复杂性。不同的组学包含不同的特征空间,可以揭示不同细胞类型的调节和功能的潜在机制,从而提供对细胞过程的全面理解。虽然最近的实验多组学策略可以测量同一细胞内的不同模式,但大量的单细胞数据由不同细胞的不成对观察组成,从而需要不匹配多组学数据分析工具。最近,一套方法展示了如何通过使用单独收集的多组学数据集来整合不匹配的数据集,包括StabMap、Cobolt、MultiVI和Seurat v5中的“Bridge Integration”。通过利用真实的多组学数据,这些方法有助于跨模态的实验设计。为了最大限度地利用来自多组学数据集的所有信息,开发包括来自输入数据的匹配和不匹配样本和/或基因的综合方法是必不可少的。尽管许多用于多组学和多层数据的计算集成工具取得了进步,但细胞类型和基因调控网络的生物功能分析仍然是一个计算挑战。 多组学数据也为识别分子模式之间的调节关系提供了独特的资源。其中包括SCENIC+,基因调控的功能推断(FigR),以及综合调控分析的概率多模态模型(MIRA),该模型通过配对转录组学和scATAC-seq测量来模拟联合变异,将增强子与基因联系起来,并识别驱动细胞决策的转录因子。这些计算方法将使多组学单细胞分析不仅提高我们对细胞分类学的理解,而且进一步加深对基因调控等基本分子过程的理解。 结论及未来展望 单细胞多组学已经成为阐明生物过程复杂性的重要方法,特别是在面对罕见疾病和细胞类型时。尽管最近取得了进展,但多组学技术需要进一步改进或探索,才能在分子细胞生物学中产生更深远和广泛的影响。例如,对新兴计算工具进行基准测试以帮助多组学研究已经变得至关重要,因为大多数多组学方法依赖于这些计算方法进行数据集成和分析。此外,很难获得表征不同组学层的匹配或相同实验数据集以及金标准数据集,这迫切需要对多组学研究进行基准测试并生成数据分析指南。 其他挑战包括测序成本高和单细胞覆盖范围有限,每个组学层只能部分分析,从而导至重要数据的丢失。为了获取更多信息和完整的基因组数据,长读技术可以推进细胞遗传变异的检测,但仍然面临测序精度低和难以从临床样本中获得完整DNA和全长RNA的限制。空间组学的出现通过提供细胞的空间坐标,从而追溯细胞在组织内的位置及其相关的多组学读数,正在改变我们对疾病背后的分子机制的理解。目前大多数空间技术在同时处理的组织数量上都是低通量的,因此不能完全捕获组织的3D结构。此外,相对较小的捕获区域限制了它们在大多数人体组织切片的完整分析中的应用。 最后,尽管提供了许多计算方法来对scRNA-seq数据进行伪时间分析,但这些方法在重建细胞发育过程的轨迹时并不总是准确的,因为细胞发育过程是复杂和动态的。因此,有必要将时间维度应用到单细胞多组学中,以统一不同数据集的多组学层。单细胞多组学领域的改进和进步将促进先进治疗策略的设计,并产生多组学和时间尺度的图谱,以帮助我们理解健康和疾病。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号