1.前言 国家药品监督管理局在2022年10.10发布了新版《医疗器械注册质量管理体系核查指南》,旧版《关于发布医疗器械注册质量管理体系核查指南的通告》(2020年第19号)同时废止。 新版指南根据《医疗器械监督管理条例》、《医疗器械注册管理办法》、《体外诊断试剂注册管理办法》、《医疗器械生产监督管理办法》、《医疗器械临床试验质量管理规范》《医疗器械注册自检管理规定》等要求制定。 指南适用于医疗器械监管部门对第二类、第三类医疗器械注册质量管理体系现场核查。2.新旧版本内容结构变化 新版《体系核查指南》相对于旧版而言,结构内容更加清晰,条款划分更细致。 新旧版本的第一章节不同,是由于2021年是医疗器械的法规大年,新版《体系核查指南》的制定依据增加了相关法规,内容增加了注册自检、委托生产、临床试验管理等新法规要求。 新旧版本的第二章一致,均适用于第二类、第三类医疗器械注册质量管理体系现场核查。 新旧版本的第三章描述不一致,但主体思想是一致的,要求企业按医疗器械GMP建立相适用的质量管理体系,要求监管机构重点关注与产品研制、生产有关的设计开发、采购、生产管理、质量控制等内容。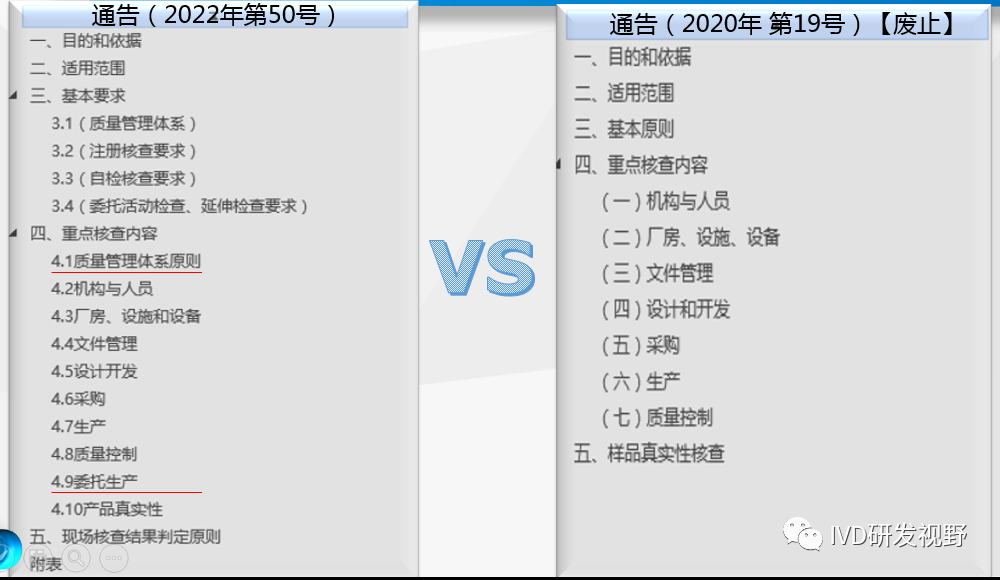
3.新版本亮点 新旧版本的第五章不一致,新版第五章及附表是最大的亮点。第五章制定了现场核查结果判定原则,现场核查结论有四种,“通过核查”、“未通过核查”、“整改后通过核查”、“整改后未通过核查”,“未通过核查”的判定原则是发现①现场检查发现申请人存在真实性问题;②现场检查未发现真实性问题,但发现申请人存在关键项目3项(含)以上或者一般项目10项(含)以上不符合要求的。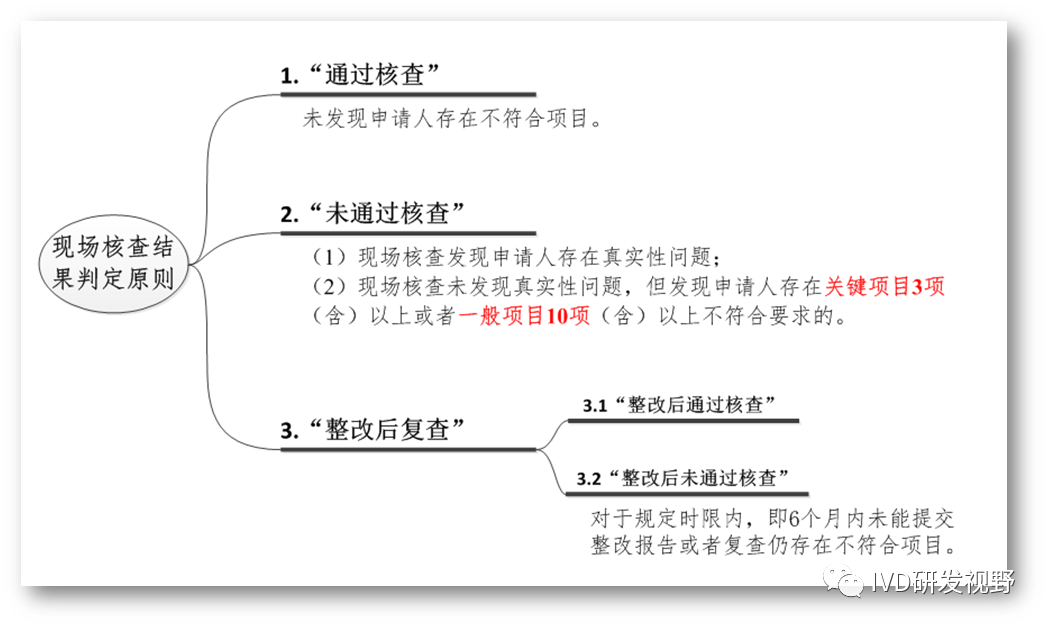
旧版核查指南里未包含关于“未通过核查”判定的介绍,判断依据是参照《食品药品监管总局关于印发医疗器械生产质量管理规范现场检查指导原则等4个指导原则的通知》规定“现场检查中发现企业关键项目(标识“*”项)不符合要求的,或虽然仅有一般项目(未标识“*”项)不符合要求,但可能对产品质量产生直接影响的,建议结论为“未通过检查”。
标准降低到了3项(含),统一了一般项不符合为10项,减轻了企业的心理负担,也给了检查员充足的判断空间。3.新版本附表 附表规定了核查的具体条款和内容,包括10个章节,(4.1质量管理体系原则、4.2机构与人员、4.3厂房、设施和设备、4.4文件管理、4.5设计开发、4.6采购、4.7生产、4.8质量控制、4.9委托生产、4.10产品真实性)共有核查项目73项,其中标注“*”关键项目32项,一般项目41项。除去委托生产和自检的规定,剩余章节关键项目的重点内容如下图。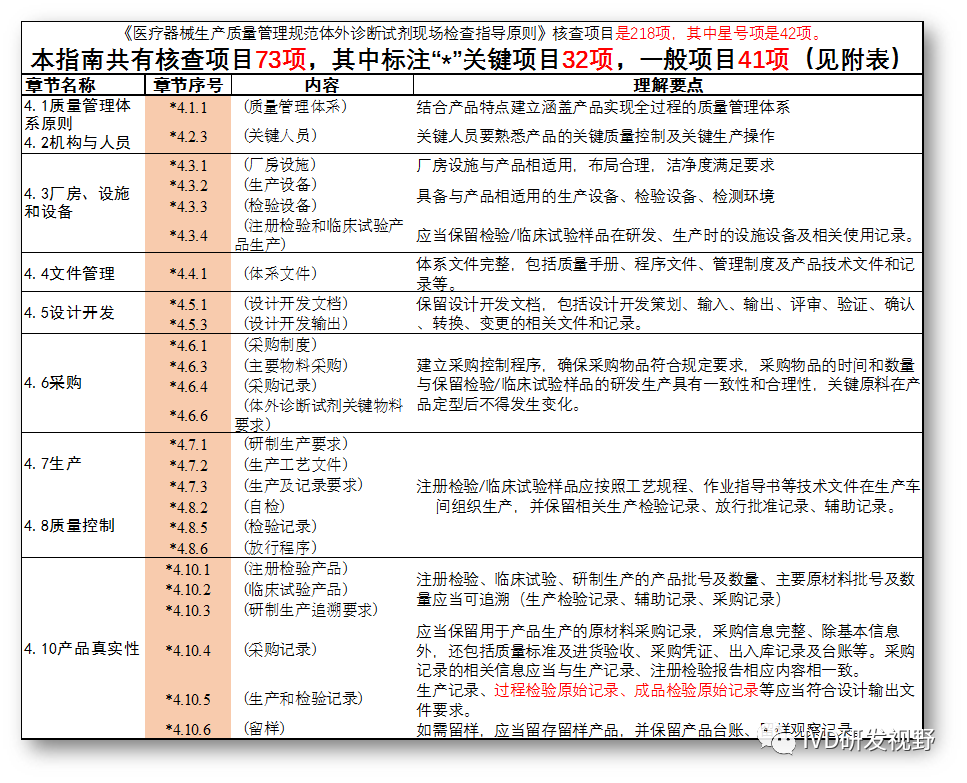
4.新版本其它新增内容 配合最新《医疗器械监督管理条例》及配套规章制度的实施,新版指南新增了自检、委托研发、委托生产、延伸检查的规定,另外还在设计开发中大篇幅增加了临床评价相关的文件管理、试验产品管理要求。
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 









