本部分内容摘自《为测试开发商提供的血清学模板》的“示例模板”部分,全文及英文原件下载,请见问文后。 J. 性能评估 应进行以下确认研究以支持你的EUA申请。请注意,特别是对于新技术,FDA可能要求额外的研究,以便我们能够充分评估与考核方法相关的已知和潜在的风险和利益。[对于每个确认研究,你应该提供一个研究方案,包括详细的、逐步的描述如何准备样品和如何进行测试。你还应包括每项确认研究的研究数据,以兼容Excel的规格提供给所有确认研究。行数据应根据测试结果的解释,介绍每个重复的最终候选血清学测试结果。如果考核方法有数字输出,你应该包括每个重复的数字值。] 1) 分析灵敏度和特异性: a) 包容性(分析反应性): 随着病毒的传播,SARS-CoV-2基因组的突变已经被发现。突变是指SARS-CoV-2病毒序列与参考序列(如Wuhan-Hu1或USA-WA1/2020)相比发生的个体遗传变化。一个新的SARS-CoV-2病毒变体有一个或多个突变,使其区别于已经在普通人群中流通的野生型或主要的病毒变体。SARS-CoV-2的变体是通过基因组序列来识别的,这些序列包含RNA基因组的突变,这可能导至病毒蛋白的氨基酸替换、插入和/或删除。不同的变体可以导至不同的表型(如,抗原性、传播性或毒性的不同)。相对于最初分离的病毒,病毒突变和病毒变体可能导至免疫原性的改变,这可能影响血清学测试的性能。如,一些研究表明,已确定的变体(可能包含多个突变)可能影响一些抗体在体外中和病毒的能力。 测试开发商应持续监测可能影响血清学测试性能的新的和正在出现的病毒突变和变体。这包括评估序列数据库(如GISAID[1]数据库)中病毒突变的流行程度,因为在这些数据库中观察到的频率很高的突变可能标志着该突变在美国的感染者中的比例越来越大。FDA目前认为显著频率大于5%(当考虑到最近一段时间内至少有2000个序列,如过去一周、一个月或一个季度)。监测还应该包括识别是否有多个可靠的报告表明某个病毒变体(可能有一个或多个突变)有可能增加毒力,增加传播,或以其他方式增加公共健康风险。鉴于突变和变种的发生率和评估其影响的重要性,FDA建议至少每月进行一次监测。 对于任何被确定为普遍存在和/或具有上述临床意义的病毒突变和变体,你应该评估由此预测的病毒蛋白中的氨基酸变化是否对你的测试设计至关重要。如果发现突变对你的测试设计至关重要,应使用临床(或伪造的,如有,适当的)样品评估突变和变体,以评估突变或变体对你测试性能的影响。 突变的总体影响不应降低测试的临床性能5%或更多,或使测试的临床性能点估计值低于J.3.d节[2]中描述的临床性能建议。 请参见FDA指导文件Policy for Evaluating Impact of Viral Mutations on COVID-19 Tests,以了解关于监测基因变异对血清学测试影响的更多讨论[3]。 FDA也有持续的监测工作,可能会确定一个病毒突变或变体具有临床意义,建议用临床(或伪造的,如果有的话,适当的)样品进行测试,以评估突变或变体对你的测试性能的影响。 [请提供你的计划,以持续监测新的和正在出现的SARS-CoV-2病毒突变和变体,并评估已被确认为流行的和/或有临床意义的突变和变体对你的测定性能的影响。] [对于在你申请EUA时作为持续监测的一部分被确认为流行和/或具有临床意义的突变和变体,请提供关于突变和变体对你的测试性能的潜在影响的信息,或解释如何充分减轻与你的器械在具有变体的个人的样品中的未知性能有关的风险。] b) 交叉反应(分析特异性): 如果大量已知的阴性样品(如,2019年12月之前在美国收集的 ≥ 75个唯一样品)从接种以下病毒疫苗和/或感染的高发人群中进行测试,并且观察到特异性 > 95%,那么目前预计不会对以下病毒进行交叉反应测试:
[如果评估的是大量已知的阴性样品(至少观察到95%的特异性),而不是上述交叉反应物的特定样品,请包括足以证明测试的样品来自于对上述病毒进行疫苗接种和/或感染的高发人群的信息,包括对测试样品的描述,如地理位置以及你所掌握的关于样品如何收集和来源的任何其他信息。] [如果大量的已知阴性样品没有被评估,请描述为评估上表中的交叉反应物而进行的交叉反应测试。请在你的描述中包括测试的样品数量和样品的制备方法]。 如果需要对交叉反应物进行测试以证明测试的交叉反应,应至少对上表所列的每种疾病/传染病原体/抗体类别的5个单独样品进行评估。 如果使用天然临床样品,重要的是使用患有基础疾病的人在感染的急性或恢复期的血清或接种特定传染病疫苗的人的血清来评估交叉反应,以获得基础疾病的高水平IgM或IgG。如果为这项研究准备了带有基础疾病IgM或IgG抗体的加标样品,那么在加样之前用考核方法确认「阴性样品」为SARS-CoV-2 IgM和IgG血清阴性是很重要的。此外,如果在COVID-19大流行之前收集的基础条件板块的市售IgM或IgG抗体可能是合适的,以确保该板块是SARS-CoV-2抗体阴性的。 我们建议你在下表中提出你的结果,并计算出考核方法结果和预期结果之间的一致性。 表 | 交叉反应:[测试名称]下面是湿测试生物体的示例 病毒/细菌/寄生虫抗体阳性
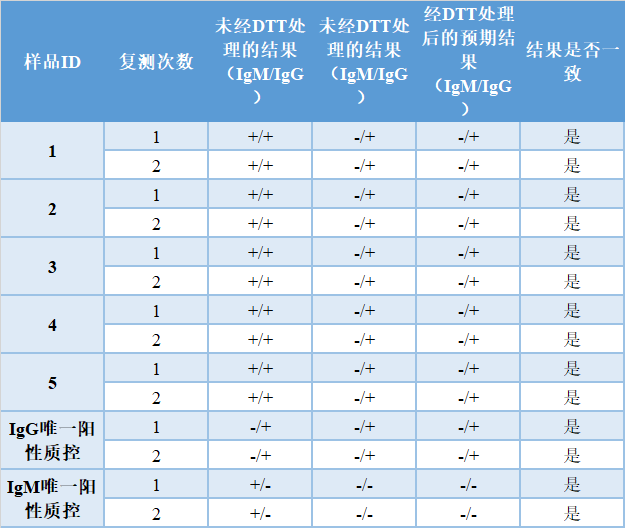
*如适用,请包括你的测试技术的输出信号值。 [如果你的测试表现出明显的交叉反应,会对所评估的任何病毒产生假阳性结果,请描述一个能充分减轻风险的计划。] 2) 不同类别的特异性(如适用) 如果你的测试旨在检测总抗,不区分不同的免疫球蛋白,那么这项研究就不适用。[在这种情况下,请说明本研究不适用]。 [如果你的测试旨在区分不同的免疫球蛋白,请描述用于评估类别特异性的方法。] 评估类别特异性的方法取决于测定规格。如果你在测试中使用了特性良好的抗IgG和抗IgM试剂,可能不需要进行类别特异性测试。在这种情况下,FDA建议描述试剂是如何被表征的,以及这种表征如何支持类别特异性。 [如果你的测试需要进行类别特异性测试,请描述所进行的一项或多项研究,以证明该测定能够准确地检测每一类抗体(如,IgG和IgM)。这应包括描述为评估人类IgM与IgG交叉反应并因此产生假阳性结果的可能性而进行的研究,以及IgM与IgG竞争并产生假阴性结果的可能性。请说明测试的样品数量,以及每个样品的重复数量。如果研究中包括纯IgG和纯IgM的阳性质控,评估至少5个样品的两个抗体类别的阳性(IgM阳性同时也是IgG阳性),一式两份,可能是合适的,如下表示例所示。本研究的商业化阳性质控是合适的(如,单克隆、重组抗体)。请提供任何类特异性测试的方案和结果,包括行数据]。 一种推荐的方法包括用二硫苏糖醇(DTT)处理样品,最终的IgG结果将保持不受影响,而最终的IgM信号将减少或为阴性。还应包括一个确认DTT活性的阳性质控。 100%与预期结果一致将确定抗体不同类别的特异性。 如果采用DTT处理方法,下面是IgM和IgG的示例表格:
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号