# 怎样看懂一份基因检测报告 # 导读 高通量测序技术也称为二代测序(Next Generation Sequencing,NGS),该技术相对一代测序大大降低了测序成本并大幅提高了测序速度,广泛应用于试管婴儿(PGD/PGS)、无创产前检查(NIPT)、遗传病的诊断、肿瘤分子诊断个体化用药等医疗场景中。 一个样本经过NGS检测后会检测出大量的变异数据,比如经全外显子测序可检测出20-40万个变异,经全基因组测序则会检测出大约400万个变异。如何从海量的数据中找到跟疾病发生发展和用药相关的基因突变呢?
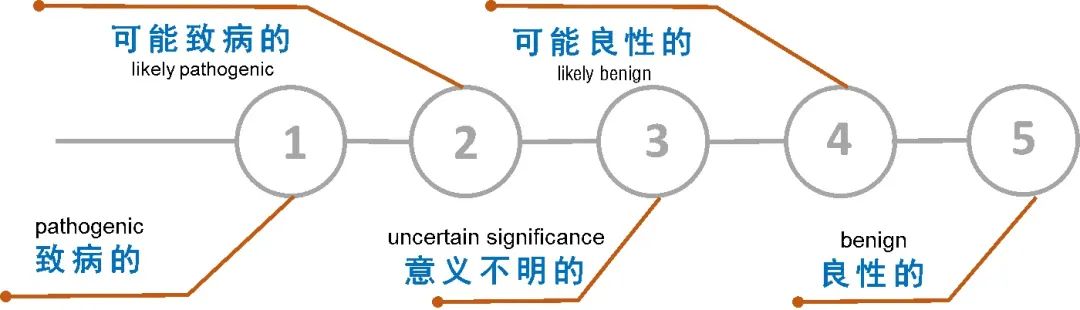
实验室产生的原始数据传给生信部门,生信分析系统通过设置一系列筛选条件把范围缩小到一定范围。最终剩下的这些变异需要我们做人工判断,美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)和分子病理协会 (the Association for Molecular Pathology,AMP)制定的ACMG/AMP指南就是帮助我们解读胚系变异的参考依据之一,同时,该指南的制定也有效地促进了实验室遗传变异分类的规范化和一致化。 ACMG/AMP指南的发展 ACMG在2013年成立了一个工作组来重新审视和修订序列变异解读的标准和指南,工作组包括ACMG、AMP和美国病理学家协会(the College of American Pathologists,CAP)的代表,该工作组依据专家建议、工作组共识和公众反馈建立了一种可以对现有的证据进行加权的系统,并应用此系统对序列变异进行标准分类。 2015年ACMG/AMP发布了最新的遗传变异分类标准与指南,该指南建议使用特定标准术语来描述孟德尔疾病相关的基因变异——“致病的(P)”、“可能致病的(LP)”、“意义不明确的(VUS)”、“可能良性的(LB)”和“良性的(B)”。
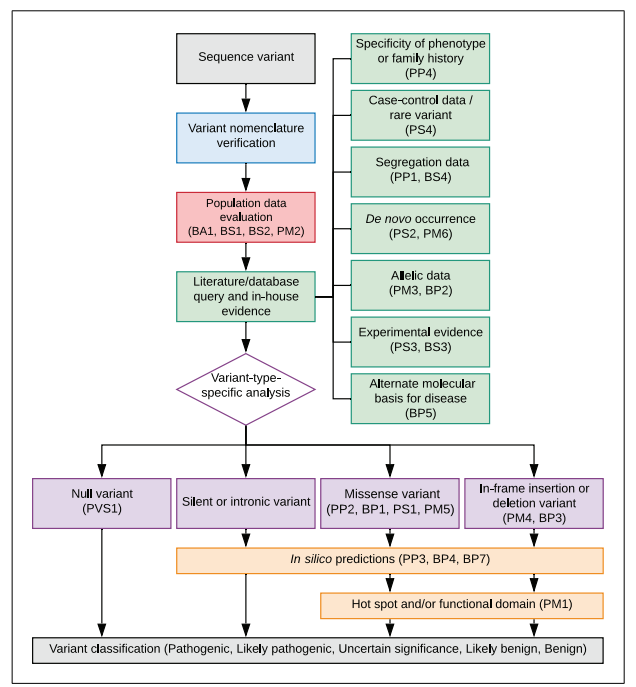
此外,该指南描述了基于典型的数据类型(如人群数据,计算数据, 功能数据,共分离数据)对变异进行五级分类的标准过程。该报告一经发表立即广泛应用于世界各地的临床实践中。随着临床信息的不断丰富,临床基因组资源(ClinGen)的序列变异解读工作组SVI和几十个疾病变异解读的专家工作组(VCEP)对指南不断地进行更新维护,对一些证据类型制定了定量化的解读指南,这些条款包括PVS1、BA1、PS3/BS3、PP5/BP6、PS2/PM6和PM3。在中国,2017年该指南经贺林院士等专家的努力翻译成了中文。2020年,上海交通大学张军玉博士联合几位专家把ACMG指南和ClinGen相关的更新整合在一起,撰写了一份变异解读的操作指南。 序列变异解读的流程 参考张军玉博士等发表的变异解读操作指南,在实际应用中解读所需的证据主要来源于4个方面:人群数据,文献报道数据,变异类型特异的证据,预测证据。将各类型的证据逐条分析,根据最新指南,每条证据可以升降级,我们在升降级时要按照指南提出的命名规则来命名。比如共分离的证据原本只给予支持性的证据强度PP1,现在,如果共分离的数量比较多,可以升到中等或者是强,那么表达就是在原来的PP1基础上加一个下划线,加一个新的证据强度Moderate/Strong。最后根据这些证据得出致病、可能致病、意义不明确的、可能良性、良性的一个分类结果。
Figure 1 Strategic planning workflow for interpretation of sequence variants。 第一类 人群数据证据 人群数据证据中有4条证据:
另外,如果有针对特定基因的变异解读指南,以上证据优先参考相应的阈值。 第二类 文献报道数据 | 内部数据 对于每篇参考文献,有以下几个方面的信息应该关注。包括但不仅限于如下: (1)临床表型及家族史(PP4)
(2)Case-control数据(PS4)
(3)共分离(PP1/BS4)
通过计算共分离的次数代入公式计算LOD值(如下图),再根据LOD值给予相应的证据等级。如遗传性耳聋变异解读指南中,当LOD值为0.6、1.2、1.5时,分别给予PP1_Supporting/ PP1_Moderate/ PP1_Strong证据等级。
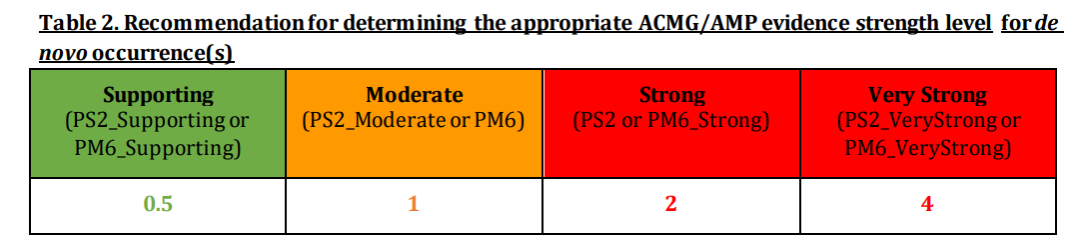
注:这部分需要考虑复杂疾病和外显率问题。 (4)新发变异(PS2/PM6)
注: 仅仅确认父母还不够, 还需注意捐卵、代孕、胚胎移植的差错等情况。
根据表型的一致性,双亲是否经过验证以及患者数量制定了新发变异得分表,然后根据得分确定新发变异发生的合适的ACMG/AMP证据强度水平:
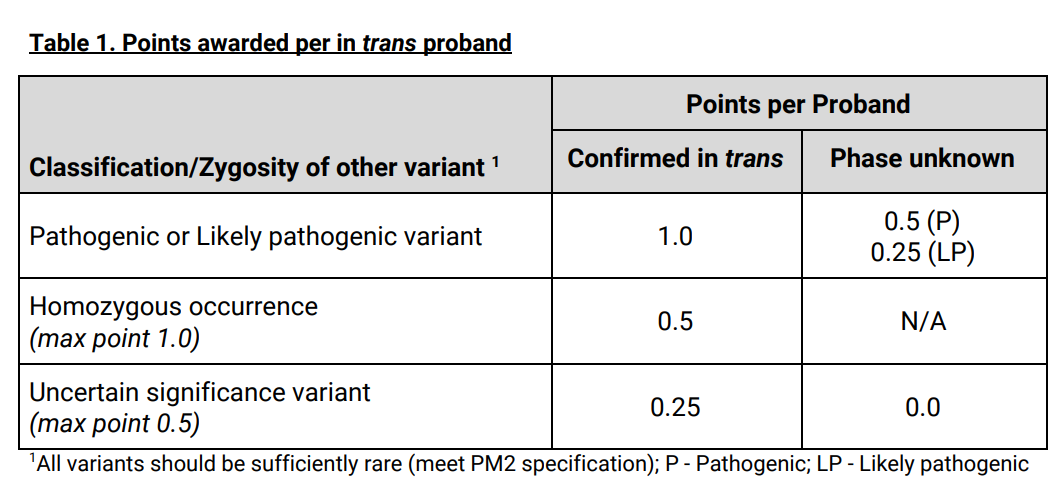
(5)等位基因数据(PM3/BP2)
根据患者中检测到的相同基因上的其他变异的分类/合子类型及相位关系制定了PM3得分表,然后根据得分将PM3的ACMG/AMP证据强度划分为:PM3_Supporting/PM3/ PM3_Strong/PM3_VeryStrong四个强度等级。该证据的使用也和PS4一样需要满足变异是非常罕见的。
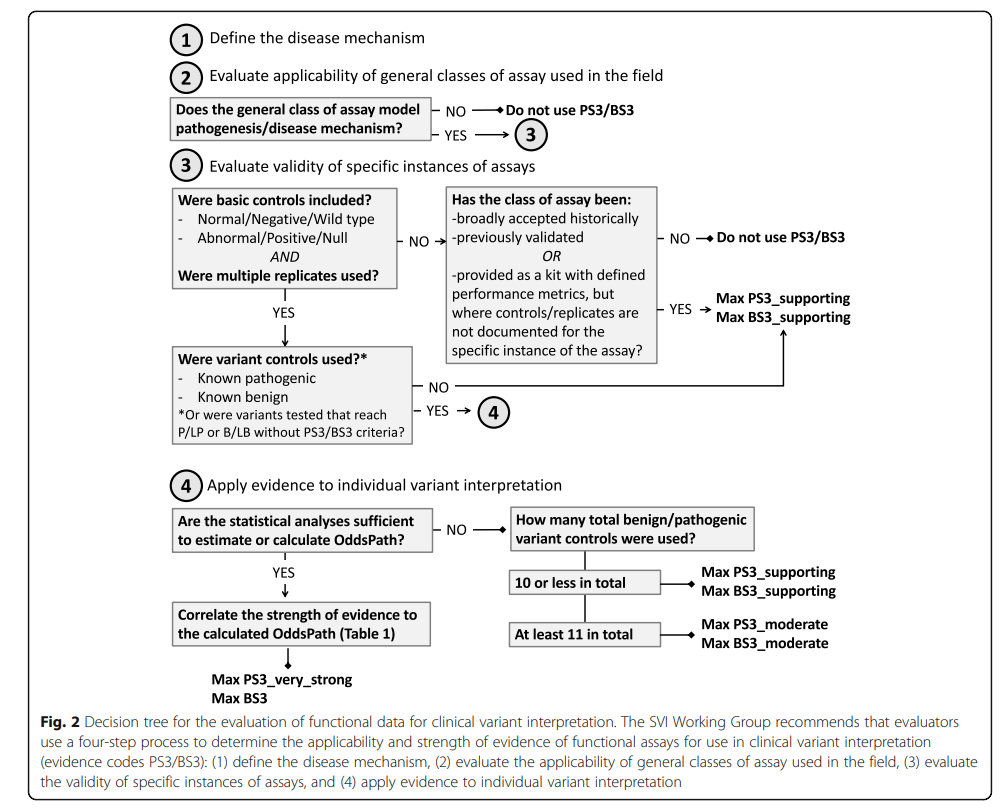
(6)功能实验(PS3/BS3)
注: 功能实验需要验证是有效的,且具有重复性与稳定性。
关于PS3的使用,ClinGen工作组介绍了PS3详细的使用流程图。我们看到功能实验的使用要求其实是非常高的,如果我们按照这个流程其实会发现论文中报道的功能试验很多只能用到PS3_Supporting的级别。
(7)其他致病变异(BP5)
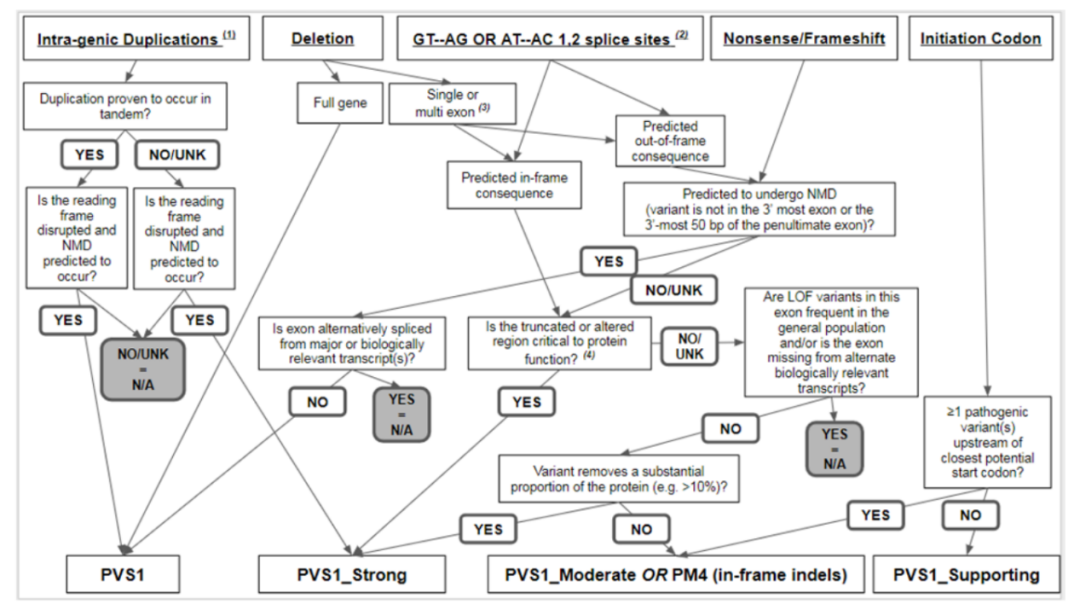
第三类 变异类型特异性证据 有的证据只适用于特定的变异类型。 (1)无功能变异(PVS1)
(2)错义突变(PS1/PM5)
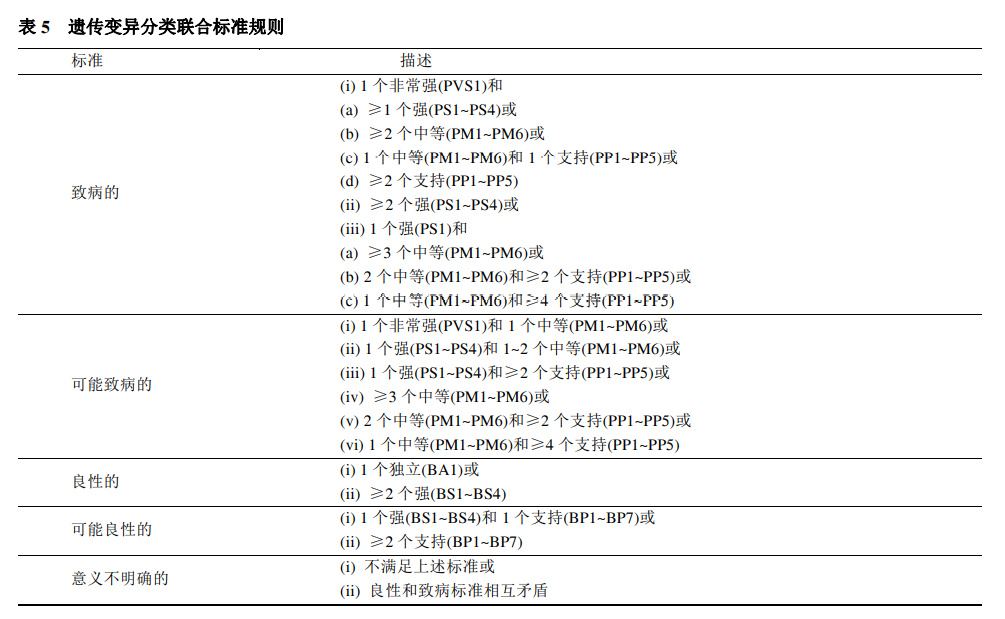
以上证据都需要注意错义变异的致病原因是否是影响剪接改变引起的。 第四类 计算机预测数据 在变异有害预测方面主要是对三个方面进行预测,分别是错义变异的预测、剪接的预测和热点和关键功能域预测。 (1)错义变异的预测(PP3/BP4) 在错义变异预测中SVI工作组建议使用REVEL作为预测工具。一般而言,REVEL>0.75时认为是符合PP3的证据,应用BP4证据的阈值目前SVI工作组还没有提出,但遗传性耳聋变异解读指南中,指出REVEL<0.15认为是符合BP4的证据。 (2)剪接的预测(BP7) 对于剪接位点预测,常用的预测软件有MaxEntScan, NNSPLICE和 HumanSplicingFinder。 (3)热点和关键功能域预测(PM1) PM1: 位于热点突变区域,和/或位于已知无良性变异的关键功能域(如酶的活性位点)。 除了已知明确的热点以及功能域之外,SVI建议错义变异或框内插入缺失变异可以根据o/e score通过比较观察到的和预期基因特定区域的错义变异数来确定关键区域。 最后,把所得的证据按照下表的标准规则分为5类。
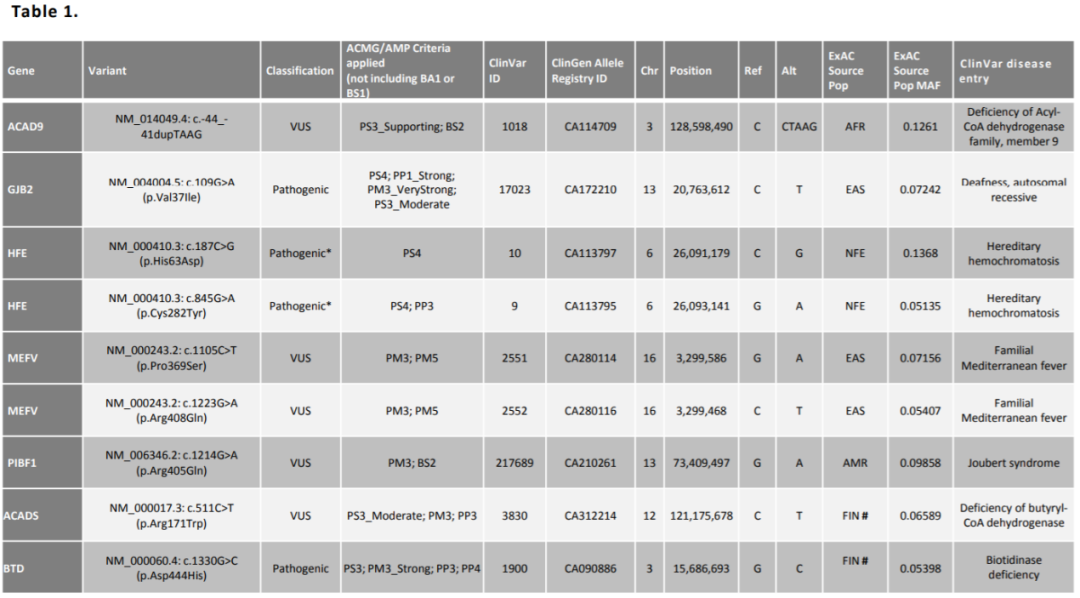
几点补充 … 这个标准仅适用于孟德尔遗传病致病基因的变异评价,这些针对变异的分类也只是针对基因变异本身来定义的,并不能直接用作疾病诊断。 基因变异即使被判读为致病,也并不代表这个变异可以作为患者疾病的遗传学诊断。 另外,意义不明的变异也不适合直接做阴性诊断,因为临床表型的判断也至关重要,只有经过临床医师对疾病表型关联和遗传特征的分析确定二者吻合,才能最终得出诊断性结论。 总结 仁东医学的报告对明确与肿瘤相关的基因进行检测分析,其中检出的胚系突变按致病、疑似致病、意义未明、疑似良性、良性进行划分,最终将重要基因的致病/疑似致病突变检测结果展示在报告中,以作为临床医生判断用药、预后及遗传分析等的重要参考依据。 下期预告 Next issue 2022年3月2日(下周三)早上8点钟,“仁东医学六周年全新企划”第十四期,由仁东医学医学部来和大家谈谈《与肿瘤相关的那些明星基因:肠癌之MRD》。敬请关注! 参考文献 … 1.王秋菊, 沈亦平, 邬玲仟, 等. 遗传变异分类标准与指南. 中国科学: 生命科学, 2017, 47: 668–688. 2.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. 3.Zhang J, Yao Y, He H, Shen J. Clinical Interpretation of Sequence Variants. Curr Protoc Hum Genet. 2020 Jun;106(1):e98. 4.ClinGen General Sequence Variant Curation Process Standard Operating Procedure |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号