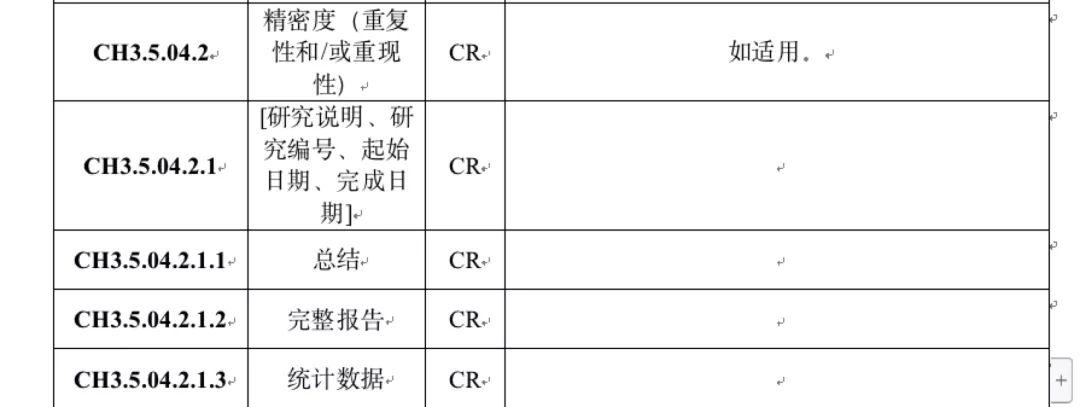
越来越多的企业加入到体外诊断试剂(IVD)行业,开始产品研发和注册申请。在产品资料技术审核阶段,会出现补正资料通知好几页纸的情况,甚至每一份技术资料都需要补充,如果补正内容涉及了注册检验和临床试验,就可能需要接近一年的时间,大大影响了企业获证的速度。 Note: 本文讲的是如何提高质量,而不是“无中生有”,注册申报资料必须是基于扎实的设计开发基础和充分的临床试验数据。 第一期: 如何撰写逻辑清楚、结构清晰的申报资料 自药监局推出医疗器械注册电子申报信息化系统(eRPS)后,申报资料的结构和要求已经更加清晰,但是很多同行依然不知道如何将内容清晰准确的表述,让审评老师快速理解。 虽然技术审评的时限是90个工作日,但是审评老师真正看资料的时间只有几天,而审评老师是第一次接触申请人的产品。如何让老师迅速了解申报的产品和提交的内容;资料的撰写逻辑和表述方式就非常重要。 重点 1) 熟悉法规要求: 研发和注册人员需要充分熟悉各类《注册申报资料指导原则》,对于每一份资料要求提供的内容了然于胸,指导审评老师需要看到哪些内容。 2) 换位思考: 把自己当作是一个不了解这个产品的人,来撰写资料。就算是一个其它厂家已经申报过的成熟产品,对于审评老师来说,申请人的产品依然是一个全新的产品,其中的内容、细节都是第一次审阅。 3)结构合理: 任何一份资料,都要先有总结,再开始详细描述;切忌一份资料从头开始一个个实验描述,每一个小结都穿插在实验数据中,最后也没有总结。 举例 以非临床研究资料中的精密度研究资料举例: 1)法规要求: 对于精密度来说,需要知道目前最低是按照CLSI EP5的要求,使用不同浓度的样本和至少三批次的试剂,进行20天以上的测试,验证产品的批内/批间、日内/日间、不同地点、不同仪器以及不同操作者之间的精密度,并按照EP5的统计方法进行计算。 2)资料结构: 从下图的eRPS目录中,就可以看到其实评审老师希望看到是总结、完整报告、统计数据三份报告;通过总结应可以完整的了解精密度研究具体做了哪些试验,数据如何,内容应至少包括: a)采用了哪几批试剂; b)对多少个样本进行了测试,样本的浓度和来源; c)测试的具体方法,是否参考了CLSI EP5的要求,进行了至少20天的测试; d)批内/批间、日内/日间、不同地点、不同仪器以及不同操作者之间的精密度计算方法、接收标准和结果。 e)结论 即使最后资料不是分成三份递交,也需要按照上述的总结—完整报告—统计数据的格式来写,因为这个格式才是最容易理解和审阅的。在总结之后,才是完整的详细报告,数据应以汇总表格的形式呈现;具体统计数据应作为附件形式后附。
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号