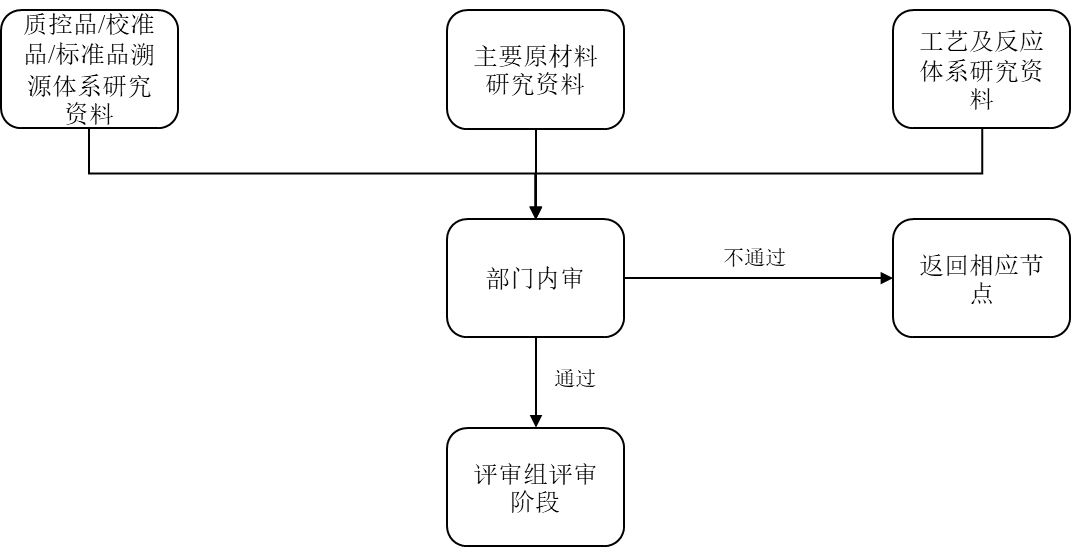
体外诊断试剂的开发 项目负责人依据研发小试开发阶段(TR4)设计开发计划书进行标本调研、主要原材料研究、工艺及反应体系研究、性能研究、稳定性研究、溯源体系研究内外包材研究等产品开发工作。 1.1 标本调研 项目负责人组织相关部门开展标本调研,标本调研涉及标本的可采集性,标本的稳定性,市场上常用的对比试剂和说明书,医院检验科医生对此项目的检需求(可包括检测时间、标本类型、加样量)等。 刘博 标本类型的选择,对于性能评估试验和后期注册临床试验方案设计非常重要。 1.2 原材料筛选 查找并筛选原材料供应商,并进行原材料研究。筛选和研究的基本原则:对研制、生产用各种主要原材料、辅料制定相应的质量标准,同时符合有关法规的要求,形成原辅料清单及采购要求。 刘博 应提供与产品质量最密切相关的原材料的选择与来源、制备过程、质量分析和质量标准等相关研究资料。若主要原材料为企业自己生产,其生产工艺必须相对稳定;如主要原材料来自市场(从其他单位购买),应提供的资料包括:对物料供应商审核的相关资料、购买合同(含价格),供货方提供的质量标准、出厂检定报告,以及该原材料到货后的质量检验资料。确定原材料的质量控制方式后,输出主要原材料研究资料。 各主要原材料筛选之后供应商应相对固定,不得随意发生变更,如需变更,应执行设计变更程序。 产品注册后,主要原材料供应商发生变化时,需要重新进行临床试验,申请许可事项变更;主要原材料发生变化时,不能申请变更注册,需按照法规进行新产品注册。 1.3 工艺及反应体系、性能研究 产品的工艺及反应体系、性能测试研究应包括检测样品的要求、检出限、特异性、准确度、精密度、线性、可报告范围、干扰物质、参考值等方面,同时还应包括参考物质及质控物研究。 刘博 性能评估需要三批试剂的研究资料。 如果试剂适用于多个机型,需要在每个适用机型上进行三批试剂的研究。 可使用中试成品进行临床样品的测试与比对试验,测试结果可作为临床试验对照试剂/方法的选择依据。 1.4 稳定性研究 稳定性研究包括实时稳定性、开瓶稳定性、冻融稳定性(如适用)、运输稳定性、机载稳定性和样品稳定性等,可根据实际需要选择合理的稳定性研究方案,稳定性决定了产品的有效期。 刘博 实时稳定性,需要进行连续三批试剂在实际贮存条件下保存至有效期后的研究资料。 1.5 溯源体系研究 项目负责人组织相关部门进行参考物质溯源调研,参考物质溯源涉及项目可用国际、国内标准物,参考物质,参考测量程序等。 有关产品溯源性的标准可以参见GB/T 21415-2008(ISO 17511)《体外诊断医疗器械一生物样品中量的测量-校准品和控制物质赋值的计量学溯源性》GB/T 21415-2008(ISO 17511)标准中规定了五种模式: 1) 具有一级参考测量程序和一级校准品、能在计量上溯源到国际单位(s)的情况; 2) 具有国际约定参考测量程序(非一级)和国际约定校准品,不能在计量上溯源至SI的情况; 3) 具有国际约定参考测量程序(非一级),无国际约定校准品,不能在计量上溯源至SI的情况; 4) 具有国际约定标准品(非一级),但无国际约定参考测量程序,不能在计量上溯源至SI的情况; 5) 具有制造商选定测量程序,但既无国际约定参考测量程序,也无国际线定校准品,不能在计量上溯源至SI的情况。 制定产品性能评定所需的质控品、校准品、标准品相关要求,输出质控品校准品/标准品溯源性研究资料。 刘博 如果没有公认的校准品和参考方法,可溯源到企业内部标准品。 体外诊断试剂评审 2.1 部门内审 项目负责人整理研发小试阶段相关技术文件,由部门组织内部评审,输出评审记录由部门主管审核通过后可归档至相关研发部门。 相关负责人负责试剂研发试样产品的溯源,为试剂研发试样的产品提供参品、工作校准品、产品校准品、质控品,并整理主要原材料研究资料、工艺反应体系研究资料、质控品校准品标准品溯源体系研究资料等,由研发部组织相部门参与评审,输出的评审记录由部门主管审核后可归档至相关研发部门。 部门内审过程见图1。
图 1 部门内评审阶段 2.2 评审组评审 部门内审通过后,项目负责人整理物料清单(BOM清单)、物料技术标准试样记录、研发小试阶段性能评估方案和报告、内包材研究资料(适用时编制外包材研究资料(适用时编制)、产品说明书、产品技术要求、产品包装标签样稿等,提交至评审组。 研发中心负责组织召开研发小试评审会议,并完成会议记录的编制、归档,评审组负责评审决议及文件的批准,相关人员负责评审记录的编制及归档,项目组根据评审结果判断该项目是否可以进入下一阶段。 刘博 项目组须在研发小试适当的时候向采购部提出新增供应商评审的需求,确保在产品转生产之前完成供应商评审,确保进入中试生产和正式生产的供应商均为合格供应商。 评审组评审过程参见下图。
图 2 评审组审评阶段 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号