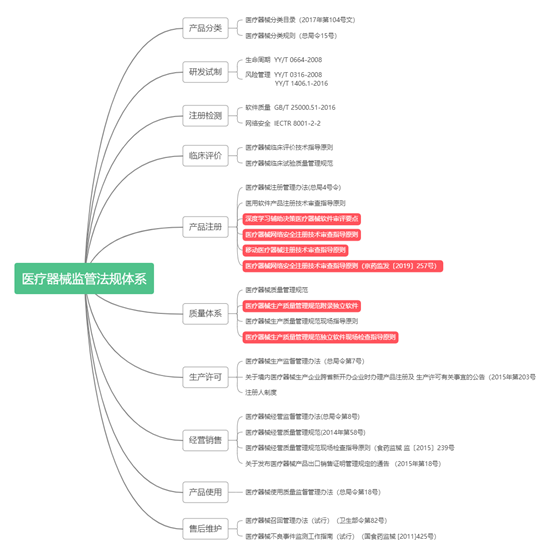
随着国内首张医疗AI产品安德医智旗下的BioMind“天医智”颅内肿瘤磁共振影像辅助诊断软件获得NMPA批准上市。对于如何进行医疗AI产品的注册上市又成为国内医疗AI行业的讨论热点,与行业内重点关注的如何进行注册不同,从医疗产品的产业化角度来看,产品是否获取批准上市,核心在于从项目立项开始,如何进行全流程质量体系的构建以及在上市注册过程中对各个环节的衔接和细节把控,尤其是全流程项目规划与管理,是产品获得NMPA批准上市的根本。 国家药监局发布了一系列规范性文件来对深度学习产品的上市注册进行指导,主要包括《深度学习辅助决策医疗器械软件审评要点》、《医疗器械软件注册技术审查指导原则》、《医疗器械网络安全注册技术审查指导原则》、《移动医疗器械注册技术审查指导原则》。根据对这些法规的分析并结合相关注册实践,下面将简要进行总结并在随后的文章中进行进一步深入思考与讨论。 1 基于深度学习算法产品的医疗器械法规体系 目前医疗器械软件公司很多都是一些创业型公司,规模小、人员背景单一。在产品研发前期没有进行产品注册策划,研发人员不懂医疗器械法规、标准。只是单纯的认为把软件研发出来交给注册人员就可以了,导至产品在检测过程中不符合法规标准要求重新更改,或者产品研发数据不充分导至重新验证确认等。反而大大浪费了企业产品上市的时间成本和费用成本。因此,在产品立项时,我们就应该关注产品的整个生命周期应适用的法规及标准,保证软件产品在各阶段的符合性。下图为医疗器械独立软件的监管法规体系图。
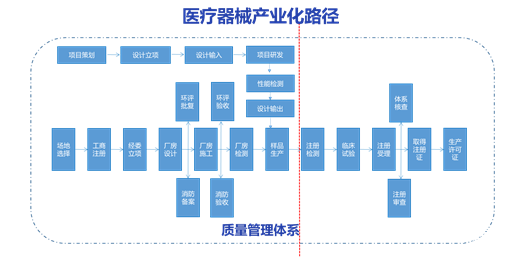
医疗器械独立软件监管法规体系 医疗器械技术审评以产品的预期用途和适用范围为出发点,最终也归结到产品的预期用途和适用范围,因此所有注册申报资料也应服务于这一核心目的。医疗器械技术审评的最终目的是向公众输出经评审过的医疗器械产品注册证、产品技术要求、产品说明书,产品说明书主要从临床应用角度对产品的预期用途和适用范围进行了详尽的描述,产品技术要求从工程技术角度对保证产品安全有效性的性能参数进行了描述,产品注册证是对企业提供的所有注册申报材料的核心内容进行描述。 除了以上几份资料外,注册资料还包括临床评价资料、研究资料、综述资料、风险评价资料、安全有效基本要求清单等主要技术资料。临床评价资料是为了证明产品可实现的预期用途和适用范围,反应产品与其应用者相关性的文件。综述资料是企业对产品的全貌进行呈现的文件,主要目的是让药监局审评人员全面的了解产品的各个细节。研究资料是企业将产品的整个研发生产过程和各种验证以及其他所有证据进行呈现的文件,同时也可以充分阐述产品的技术要求的产生全过程,说明产品技术要求的来之不易及其与产品实际完全贴切。风险评价贯穿产品全生命周期,风险评价资料是推动产品全生命周期产品质量提升的动力源,也是反映体系控制和由其诞生的注册资料两者间的重要桥梁文件。安全有效基本要求清单是在开发活动结束后,申报注册前,充分梳理的企业围绕产品开发的全部思维活动、所借鉴的外部资源和已完成的全部活动的文件。 2 基于深度学习算法产品的产业化路径与质量体系建立 根据《医疗器械监督管理条例》和《医疗器械注册管理办法》,在质量体系的管控下,医疗器械产品在完成研发产品定型后,进一步完成注册检测和临床评价后可以向NMPA申请注册受理。在申请注册所准备的资料中,在企业完整的体系框架下,综述资料和研究资料均是可以在产品设计文件和验证文件中获取,因此在医疗器械产品的产业化中,申报资料中的综述资料和研究资料的准备并不是根据法规的要求进行起草,而是应诞生于现有的体系控制。如下面的流程图所示,产品进入市场是研发项目进度和相应的生产体系并行推进的过程,二者的发展进度相互制约影响和协同推动。 《医疗器械监督管理条例》和《医疗器械注册管理办法》,在质量体系的管控下,医疗器械产品在完成研发产品定型后,进一步完成注册检测和临床评价后可以向NMPA申请注册受理。在申请注册所准备的资料中,在企业完整的体系框架下,综述资料和研究资料均是可以在产品设计文件和验证文件中获取,因此在医疗器械产品的产业化中,申报资料中的综述资料和研究资料的准备并不是根据法规的要求进行起草,而是应诞生于现有的体系控制。如下面的流程图所示,产品进入市场是研发项目进度和相应的生产体系并行推进的过程,二者的发展进度相互制约影响和协同推动。
医疗器械软件包括独立软件与软件组件,注册检测和临床评价是软件产品产业化的必经之路,也是绝大多数企业的关注点,但医疗器械设计开发的重要需求输入是产品技术要求和需求规范,注册检测则是对产品技术要求需求输入的系统验证,临床试验则是对产品需求输入中临床部分要求的系统验证,因此产品获取上市认证的路径要从产品的项目立项开始,并且在一个良好质量体系运行的基础上来完成。
3 产品分类 依据《医疗器械分类规则》、《医疗器械分类目录》,医疗器械软件分为独立软件与软件组件,我们从医疗器械定义;产品的预期用途、使用场景、核心功能;辅助决策的程度三个方面进行分类判定。 3.1产品的预期用途、使用场景、核心功能 产品预期用途:主要考虑软件的临床用途(如诊断、治疗、监护、筛查等)和重要程度(如重用作用、辅助作用、补充作用等)。 使用场景:主要考虑软件的使用场所(如医院、家庭等)、疾病类型(如严重性、紧迫性、传染性等)、患者人群(如成人、儿童、老年、女性等)和用户类型(如专业用户、普通用户、患者等); 核心功能:包括功能类型(如控制驱动、处理分析等)、实现方法(如CT图像重建采用滤波反投影算法还是迭代算法,异常识别采用常规图像处理算法还是人工智能算法等)和复杂程度(如算法规模、参数数量、运算速度等) 3.2辅助决策程度 诊断功能软件风险程度按照其采用算法的风险程度、成熟程度、公开程度等为判定依据,不仅仅依据处理对象(如:癌症、恶性肿瘤等疾病的影像)为判定依据。若诊断软件通过其算法,提供诊断建议,仅具有辅助诊断功能,不直接给出诊断结论,本子目录中相关产品按照第二类医疗器械管理。若诊断软件通过其算法(例如,CAD,骨密度除外)对病变部位进行自动识别,并提供明确的诊断提示,则其风险级别相对较高,本子目录中相关产品按照第三类医疗器械管理。
综上所述,产品分类不是简单的以产品名称或软件算法类型进行分类,而是综合产品预期用途、应用场景、核心功能等因素进行综合分类判定。同时企业也要咨询医疗器械审评中心,或进行分类界定最终确定。 我们找了近几年的软件产品的分类界定结果示例供大家参考:
4 命名规则 4.1医疗器械通用名称由一个核心词和一般不超过三个特征词组成
4.2辅助决策软件术语说明:
举例:如安德科技的产品名称为,“颅内肿瘤磁共振影像辅助诊断软件”。 产品核心词为“辅助诊断软件”,特征词1:颅内肿瘤;特征词2:磁共振影像。 4.3在软件产品命名中应注意以下几点:
5 软件版本 软件的命名规则一般是:V(主版本).(子版本).(修正版本).(发布日期) 软件版本分为发布版本和完整版本,其中发布版本就是主版本,即V1.0/V1;完整版本即V1.0.0.0000。 当主版本发生变化应进行许可事项变更,子版本和修改版本发生变化无需进行注册变更,按企业内部产品设计开发控制程序进行质量控制,具体版本号变更原则如下:
‐ 临床功能模块有大的变动,核心算法发生显著改变或替换;软件输出结果改变,用户使用习惯改变(界面布局改变),影响到患者安全的改变; ‐ 软件运行平台跨越互不兼容的计算平台(包括硬件和软件); ‐ 软件的安全性级别、体系结构、用户界面关系或物理拓扑发生改变
注:在产品说明书和技术要求中不必体现完整版本。 综上所述,基于深度学习算法的医疗器械软件的注册上市不仅仅是简单的注册检测和临床评价以及相应的注册资料的准备,更重要是在产品全生命周期内,以产品的研发和生产为基础,对企业“人”“财”“物”的资源调配和相应的体系控制,本文后续将持续对产品全生命周期的产业化路径进行分析和解读。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号