由于伴随诊断试剂的高风险特性及其重要的临床价值,各国监管部门均积极制定相关政策助推伴随诊断试剂行业的健康发展。随着相关监管政策的不断完善,伴随诊断试剂在精准医学时代将发挥更大的作用。
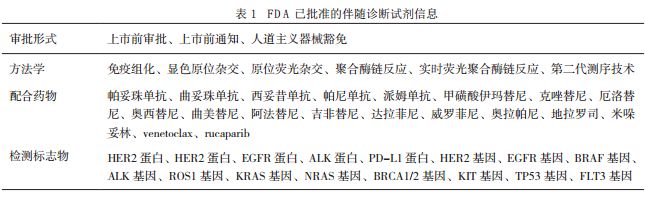
文|刘东来,王佑春,张春涛(中国食品药品检定研究院) 由于伴随诊断试剂的高风险特性及其重要的临床价值,各国监管部门均积极制定相关政策助推伴随诊断试剂行业的健康发展。随着相关监管政策的不断完善,伴随诊断试剂在精准医学时代将发挥更大的作用。 伴随诊断试剂(companion diagnostics,CDx)是一类能够为患者提供特定药物应用的安全性与有效性信息的体外诊断试剂,能帮助医疗工作者评估不同患者的受益程度以及潜在副作用与风险,并确定最终治疗方案[1]。伴随诊断试剂的出现和发展,与精准医学和个体化治疗药物的发展密不可分。1998年,美国食品药品管理局(U.S. Food And DrugAdministration,FDA)批准了赫赛汀(herceptin)用于治疗阳性转移性乳腺癌[2]。赫赛汀的有效成分是曲妥珠单抗(trastuzumab),能与人体表皮生长因子竞争结合癌细胞表面的2型人体表皮生长因子受体(human epidermal growth factor receptor 2,HER2),从而阻断癌细胞生长并激活自身免疫系统杀伤癌细胞。随赫赛汀一同获批的诊断试剂,还有达科公司(DAKO)用于检测乳腺癌细胞中HER2蛋白表达水平的体外诊断试剂。 精准医学的基础是精准诊断:一是筛选适合接受特定药物治疗的患者,二是筛选不适合接受特定药物治疗,即可能产生严重副作用与较高风险的患者,三是监测患者的治疗效果并指导制定最适合患者的治疗方案,这三点正是伴随诊断具备的特点[3]。与传统体外诊断试剂相比,伴随诊断试剂一旦发生误诊不仅将延误患者治疗,甚至可能产生严重的副作用危及患者生命。因此,伴随诊断试剂的高风险特性决定其必须接受更严格的监管。此外,药物研发领域一直以来都采用低成功率的“试错法(trial and error)”,不仅使药企研发成本居高不下,临床试验提供者与参与者也要承担很大风险[4]。伴随诊断试剂,特别是分子类伴随诊断试剂与个体化基因靶向性药物的联合开发,能有效地降低研发成本、提高研发效率并控制临床试验风险。 伴随诊断试剂对患者和药企都具有重要意义,因此,各国的监管政策一直备受关注。美国是第一个提出伴随诊断试剂概念的国家,也是第一个制定并实施针对性监管政策的国家[5]。因此,美国的监管经验具有重要参考价值。近年来,欧盟和日本的监管部门也继美国之后引入伴随诊断试剂定义,并正在制定针对性监管政策[6]。我国现阶段尚未引入伴随诊断试剂定义,也未制定针对性监管政策[7]。本文针对美国伴随诊断试剂的发展、伴随诊断试剂监管政策的发展、伴随诊断试剂指导原则、伴随诊断试剂面临的监管挑战以及各国伴随诊断试剂的监管现状等五个方面进行简要介绍,希望能为我国体外诊断试剂从业者及监管人员提供相关参考信息,并推动我国伴随诊断试剂相关产品的研发以及监管政策的制定。 截至2017年6月29日,FDA已经批准针对19种药物的34个伴随诊断试剂,其相关信息详见表1。除了1个软件类伴随诊断试剂按照“上市前通知[510(k) Premarket Notification,510(k)]”程序审批,其余33个试剂中31个经过最严格的“上市前审批(Premarket Approval,PMA)”程序进行审批,2个按照“人道主义器械豁免(Humanitarian Device Exemption,HDE)”程序审批。这些伴随诊断试剂的预期用途均被严格地限制为临床用药指导,检测方法学包括免疫组化(immunohistochemical,IHC)、显色原位杂交(chromogenic in situ hybridization,CISH)、原位荧光杂交(fluorescence in situ hybridization,FISH)、聚合酶链反应(polymerase chain reaction,PCR)、实时荧光聚合酶链反应(real-time polymerase chain reaction,RT-PCR)以及第二代测序技术(nextgeneration sequencing,NGS)等。这些伴随诊断试剂中,除了1个软件类和4个免疫类诊断试剂以外,其余29个均为分子类诊断试剂,可以说伴随诊断试剂的发展与分子诊断技术的发展密不可分。特别值得关注的是FDA于2016年10月19日、2017年6月22日和29日先后批准了3个分别针对卵巢癌、肺癌和胃癌临床指导用药的NGS伴随诊断试剂,表明NGS技术的发展已经得到权威监管部门的认可。
美国是最早提出伴随诊断试剂概念并对其进行针对性监管的国家,并在伴随诊断试剂监管发展的早期扮演引领者的角色[8]。美国承担伴随诊断试剂监管职责的主要单位均为FDA下属单位,分别是器械和辐射健康中心(Center for Devices and Radiological Health,CDRH)、生物制品评价和研究中心(Center for Biologics Evaluation and Research,CBER)、药品评价和研究中心(Center for Drug Evaluation and Research,CDER)以及国家毒理学研究中心(National Center for Toxicological Research,NCTR)。 2002年,CDER开展“基因组数据自愿捐献计划”并于医药行业协会进行一系列关于药物基因组学的研讨会;CDRH成立体外诊断试剂安全评价办公室;NCTR建立了生物信息学卓越中心、功能基因组学中心及结构基因组学中心。这些举措表明FDA已经重视个体化药物和伴随诊断试剂的监管问题并开始布局监管策略的探索。2003年,“人类基因组计划”宣布提前完成,这一里程碑事件在众多领域产生了难以估量的影响[2]。在医疗领域,基因信息与疑难疾病之间的关系开始广受关注,药物基因组学深刻改变了新药研发的进程,也推动了测序技术与生物信息学分析技术的快速发展。更多类似曲妥珠单抗与生物标志物HER2之间的关系被发现,更多的个体化药物被推向市场,如帕妥珠单抗(perjeta)、西妥昔单抗(cetuximab)等[9]。 随着个体化药物不断出现,FDA对个体化药物和伴随诊断试剂的监管探索也进一步深入。2004年,CDER的临床药理学办公室内部成立了基因组学与靶向性治疗小组,致力于基因靶向性药物;CDRH的科学技术办公室并入科学与工程实验室。2006年,NCTR的药物基因组学与分子流行病学部并入新成立的个体化营养与医学部。2009年,CDRH设立个体化医学职位,CBER设立内部研究科学家联盟。2010年,CBER组建基因组学安全性评估团队。2011年,CDRH、CBER、CDER及医疗产品和烟草办公室四部门联合建立药物与诊断国际合作中心;CBER成立个体化医学部;CDRH、CBER及CDER三部门联合发布《体外伴随诊断试剂指导原则草案》,这是国际上第一个针对伴随诊断试剂监管的指导原则。2012年,NCTR的系统生物学部门重新组建三个分支小组,分别是生物标志物与替代模型小组、创新性安全与技术小组以及个体化医学小组。此后,FDA的监管政策持续稳步向前推进。 美国是基因靶向性检测开展最早、应用最广泛、专利数量最多并且市场化程度最高的国家,因此在伴随诊断试剂领域积累的监管经验值得参考[10]。在集合多个下属单位的意见并与行业从业者充分讨论后,FDA于2014年和2016年先后发布《体外伴随诊断试剂指导原则》《体外伴随诊断试剂与治疗药物联合开发指导原则草案》[1, 11]。两份指导原则对“个体化药物-伴随诊断试剂”联合研发方案的设计、执行和评估等方面提出了建议,旨在促进与加快联合开发进程。 《体外伴随诊断试剂指导原则》于2011年7月首次发布草稿版,2014年8月发布最终版。指导原则的主要目标从业者包括:一是计划研发个体化药物(新产品或针对新适应证的已有产品)的药企,其研发的新药的安全性与有效性依赖伴随诊断试剂进行指导;二是计划研发伴随诊断试剂用于指导个体化用药的体外诊断试剂企业。指导原则的内容从伴随诊断试剂的背景、伴随诊断试剂的定义与用途、个体化药物及其伴随诊断试剂的审查及批准、研究性应用以及伴随诊断试剂说明书等五个方面提出监管意见,旨在实现以下目的:第一,明确伴随诊断试剂的定义;第二,阐述FDA监管伴随诊断试剂的理由及意义;第三,正式阐明FDA的立场,即在大多数情况下,个体化药物及其伴随诊断试剂标识的预期用途应同步获批;第四,为行业从业者与监管人员提供上市前监管途径与FDA强制监管措施等方面的指导性意见;第五,阐述当个体化药物需要标识对其使用安全性与有效性极为关键的伴随诊断试剂时,应当遵循的法定审批要求与监管部门的建议。 2016年7月,FDA发布了覆盖范围更广、更详细的《体外伴随诊断试剂与治疗药物联合开发指导原则草案》并公开征集反馈意见。新指导原则在前一个指导原则的基础上,更侧重对“个体化药物-伴随诊断试剂”联合研发的实践应用提供具体的指导性意见,内容涉及联合研发与同步获批的通用原则,具体执行临床试验过程中的具体监管要求,以及提交“个体化药物-伴随诊断试剂”申请过程中的行政流程与注意事项。具体来讲,新指导原则的内容覆盖,包括通则、研究性体外诊断试剂与药物的监管、“个体化药物-伴随诊断试剂”联合研发的前期规划、药物临床试验设计的注意事项、关于药物研发末期的体外诊断试剂研发的注意事项、同步上市批准的注意事项、说明书注意事项以及上市后监管。此外,新指导原则额外增加四个附录,分别是联合研发流程中的关键问题、样本处理注意事项、上市前审批所需的生物研究检测信息以及批准信。 伴随诊断试剂监管的挑战主要来源于技术与监管模式两方面的挑战。从“人类基因组计划”完成至今,药物基因组学与诊断学技术取得了爆发性发展,造成了企业与监管部门之间科技水平的不对称。新药研发企业与体外诊断试剂企业倾向将前沿科技转化成产品,而监管部门的科技水平发展往往滞后于企业。不仅如此,企业与监管部门对前沿技术理解的角度不同,也是监管面临的挑战之一[12]。此外,基于前沿技术的创新性产品可能需要多个监管部门联合监管,因此,更高效的监管模式可能需要对传统模式进行创新。 由于第二代测序技术快速发展并普及,科学家仅用短短几年时间就获得海量的基因数据,在药物基因组学与诊断学领域产生了革命性影响。然而,如何从海量数据中筛选有效的信息来加快产品转化是所有企业必须面临的问题。大量覆盖广泛且指向明确的基因数据能帮助科学家揭示疾病的发病机理与发展机制,还能有效地发现新的生物标志物并评估其风险指示作用。这些研究成果对新药研发至关重要,同时也对评价以新生物标志物为靶向的诊断试剂的分析性能至关重要。另一方面,如何从企业提供的海量数据中筛选有效的信息,验证产品的临床有效性与安全性,是监管部门需要解决的问题。尽管面临诸多挑战,但仍然可以预见,越来越多的企业倾向选择“个体化药物-伴随诊断试剂”联合研发的模式,有助于提高新药筛选与临床试验的效率。 为应对伴随诊断试剂的监管挑战,2010年,FDA宣布“监管科学项目”,展现了在前沿科技成果转化这一过程中扮演更重要角色的决心。该项目的核心内容是通过鼓励临床评价与个体化药物的创新来推动药物研发速度[13]。此外,FDA还通过开展一系列研究项目支持监管模式创新。这些研究项目包括:“生物标志物鉴定项目”对CDER的工作提供技术支持;“微阵列芯片与测序质量控制计划”对基于新技术的诊断试剂及其配套新标准的制定提供技术支持;“全基因组测序评价用参考基因文库项目”,有助于监管部门对第二代测序技术在个体化药物与伴随诊断试剂联合研发过程中的应用提出合理的指导意见;“第二代测序分析用高性能集成虚拟环境”是CBER开展的项目,帮助监管人员进行相关研究分析。此外,还有高分辨率人白细胞抗原分型研究项目、分子血型分析技术研究以及新型临床试验设计与方法学研究等,这些研究项目与计划将帮助监管部门更有效地应对伴随诊断试剂面临的技术挑战。 2013年,日本和欧盟继美国之后也引入伴随诊断试剂定义,并正在积极制定针对性监管政策[6, 14]。从2013年欧盟议会对伴随诊断试剂定义的修正案到2015年欧盟新版体外诊断试剂法规里面对伴随诊断试剂的定义,欧盟的监管政策不断进行调整。根据欧盟现行的体外诊断试剂指令,并没有伴随诊断试剂的定义,而欧盟内部各成员对伴随诊断试剂的定义也有不同看法。根据欧盟理事会的提案,新的体外诊断试剂法规中,对伴随诊断试剂的定义是配合个体化药物使用,用以保证药物使用的安全性与有效性,能预测患者使用该治疗药物的受益程度或风险系数,还能监测治疗有效性并指导治疗方案调整的体外诊断试剂。因此,欧盟与美国关于伴随诊断试剂的监管政策比较相近。同时,新体外诊断试剂法规也促使欧盟与美国的监管部门开展更密切的合作,共同推动伴随诊断试剂行业的发展。 日本负责伴随诊断试剂监管的部门包括药品与医疗器械局(Pharmaceuticals and Medical Devices Agency,PMDA)与健康、劳工及福利部(Ministry of Health, Labor and Welfare,MHLW)。2013年,PMDA发布《伴随诊断试剂与药物申请审批通知》,通知的内容包括伴随诊断试剂的定义以及“个体化药物-伴随诊断试剂”联合研发的注意事项等[15]。与美国相比,日本监管政策主要有两点不同。第一个不同点是,FDA建议个体化药物及其伴随诊断试剂应同步获得批准,而PMDA建议个体化药物及其伴随诊断试剂应同步提交申请。相比同步获批,要做到同步申请需要药企与体外诊断试剂企业在研发早期就开展密切合作,这种早期合作又会促进伴随诊断试剂先于个体化药物获批。第二个不同点是,如果某种需要配合伴随诊断试剂使用的药物被用于治疗严重威胁生命的疾病时,FDA认为可以在没有相应伴随诊断试剂的情况下先行批准药物。与之相反的,PMDA特别指出当伴随诊断试剂的审批如遇到不可避免的延期时,是否批准与其配套的药物时必须具体问题具体分析再做决定。 相比美国、欧盟和日本,目前我国尚未引入伴随诊断试剂定义。伴随诊断试剂在我国仍按照体外诊断试剂进行注册管理,应符合2014年7月发布的《医疗器械注册管理办法》《体外诊断试剂注册管理办法》要求[16-17]。根据《体外诊断试剂注册管理办法》中第三章和第十七条以及《6840体外诊断试剂分类目录(2013版)》中的有关规定,肿瘤相关体外诊断试剂按第三类体外诊断试剂管理,其它部分伴随诊断试剂类产品可归属第二类体外诊断试剂[17-18]。伴随诊断试剂类产品的临床试验应符合《体外诊断试剂临床研究技术指导原则》要求[19]。此外,作为国家食品药品监督管理总局的技术支撑单位,中国食品药品检定研究院于2016年新成立了体外诊断试剂检定所,显示了我国监管部门对体外诊断试剂行业加强监管的态度和决心。 精准医学时代的到来,使个体化或可定制化治疗药物或治疗方案成为可能,而伴随诊断试剂提供的精准诊断结果是这一切的基础与前提。由于个体化药物或治疗方案的安全性与有效性严重依赖伴随诊断试剂的检测结果,因此伴随诊断试剂相比传统体外诊断试剂具有更高的使用风险。此外,伴随诊断试剂能加快新的个体化药物研发流程、提高临床试验效率、缩短临床试验时间以及降低药企研发成本。因此,伴随诊断试剂对药企与体外诊断试剂企业具有重要意义。由于伴随诊断试剂的高风险特性及其重要的临床价值,各国监管部门均积极制定相关政策助推伴随诊断试剂行业的健康发展。随着相关监管政策的不断完善,伴随诊断试剂在精准医学时代将发挥更大的作用。 来源:中国药事 参考文献: [1] U.S. Food and Drug Administration. Guidance for Industry and Food and Drug Administration Staff: In Vitro Companion Diagnostic Devices [S]. 2014. [2] U.S. Food and Drug Administration. Paving the Way for Personalized Medicine: FDA's Role in a New Era of Medical Product Development [S]. 2013. [3] U.S. Food and Drug Administration. Companion Diagnostics [S]. 2016. [4] Spear B, Heath M, Huff J. Clinical Application of pharmacogenetics [J]. Trends in Molecular Medicine,2001,7(5):201-204. [5] Mansfield A. FDA Perspective on Companion Diagnostics:an Evolving Paradigm [J]. Clinical Cancer Research An Official Journal of the American Association for Cancer Research,2014,20(6):1453-1457. [6] Nagai S, Urata M, Sato H, et al. Evolving Japanese Regulations on Companion Diagnostics [J]. Nature Biotechnology,2016,34(2):141. [7] 韩昭昭. 伴随诊断类体外诊断试剂的临床试验研究浅析 [J]. 诊断病理学杂志,2015,22(7):447-448. [8] Agarwal A, Ressler D, Snyder G. The Current and Future State of Companion Diagnostics [J]. Pharmacogenomics and Personalized Medicine,2015,8:99-110. [9] U.S. Food and Drug Administration. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools) [S]. 2016. [10] 刘东来,石大伟,张春涛. 美国对实验室研发诊断试剂的监管之路 [J]. 中国新药杂志,2016,(3):244-252. [11] U.S. Food and Drug Administration. Draft Guidance for Industry and Food and Drug Administration Staff:Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product [S]. 2016. [12] Philip R, Carrington L, Chan M. US FDA Perspective on Challenges in Co-developing in vitro Companion Diagnostics and Targeted Cancer Therapeutics [J].Bioanalysis,2011,3(4):383-389. [13] Yu T, Li Q, Gray G, et al. Statistical Innovations in Diagnostic Device Evaluation [J]. Journal of Biopharmaceutical Statistics,2016,(11):1067-1077. [14] Senderowicz A, Pfaff O. Similarities and Differences in the Oncology Drug Approval Process between FDA and European Union with Emphasis on in vitro Companion Diagnostics [J]. Clinical Cancer Research An Official Journal of the American Association for Cancer Research,2014,20(6):1445-1452. [15] Pharmaceuticals and Medical Devices Agency. Notification on Approval Application for in vitro Companion Diagnostics and Corresponding Therapeutic Products [S]. 2013. [16] 国家食品药品监督管理总局. 医疗器械注册管理办法[S]. 2014. [17] 国家食品药品监督管理总局. 体外诊断试剂注册管理办法 [S]. 2014. [18] 国家食品药品监督管理总局. 体外诊断试剂分类子目录(2013版) [S]. 2013. [19] 国家食品药品监督管理总局. 体外诊断试剂临床试验技术指导原则 [S]. 2014. |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号