内容提要:通过对美国FDA、欧盟、日本等国家和地区医疗器械注册审评模式的研究,结合我国国情和现有法规模式,提出医疗器械注册审评项目管理制度,规范医疗器械注册审评过程,保证医疗器械注册审评科学、高效地开展。 关 键 词: 医疗器械注册 审评 项目管理 2017 年10 月,中共中央办公厅、国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,进一步提出完善技术审评制度。研究并践行医疗器械注册审评项目管理制度已成为深化审评审批制度改革工作的重要组成部分,并在医疗器械技术审评管理体系中占有核心地位。本文通过借鉴美国、欧盟、日本等国家和地区在审评模式的现状和经验,为我国医疗器械注册审评项目管理制度的建立提供参考。 1.美国CDRH 的审评模式研究 设备仪器与放射健康中心(Center for Devices and Radiological Health,CDRH)按风险等级对医疗器械实行分类管理(见图1),并依据产品风险进行豁免上市前通告(510(k))、上市前通告(510(k))或者上市前批准(PMA)三种不同形式的上市前审评模式,其中PMA采取项目管理的方法,因此对PMA的项目管理方法进行研究。
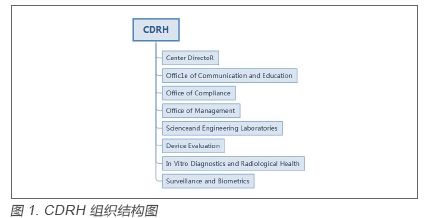
1.1 CDRH 的组织架构 CDRH分为八个部门,其中器械评估办公室(Office of Device Evaluation,ODE)和体外诊断及放射健康办公室(Office of In Vitro Diagnostics and Radiological Health,OIR)负责医疗器械产品的上市前审评,ODE以临床专业不同下设7 个业务部门(Division),每个业务部门又按照品种临床用途的不同划分不同的科室(Branch),每个科室按照产品的分类归属审评对应的品种。见图2,3。
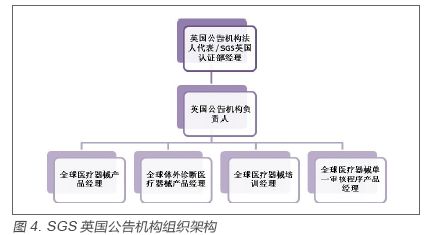
1.2 PMA 的审评流程 上市前批准(Pre-market Approval,PMA)的审评大致分为受理审评、立项审评、实质审评、专家组审评等过程。 1.2.1 受理审评 受理审评是依据受理审查问题清单,评估申请的完整性。申请人将资料提交给CDRH的文件控制中心(Document Control Center,DCC),DCC确认申请人已经支付相应费用且已经收到经验证的电子申报资料后,将申请资料发送给适当的ODE或OIR部,ODE或OIR部收到申请文件后立即转入相应的科室(Bruch),科室指派一名主审(Lead Reviewer),该主审人员在15 日内完成受理审评。受理审评的决定应由主审在直属上级(Bruch Chief)同意下作出。 1.2.2 立项审评 立项审评是依据立项审查问题清单,对基本技术要素进行审评。立项审评需要制定立项审评时间表;建立审评项目组,组织立项审评会议;制定资料的跟踪、分发和处理的流程,立项审评的决定应由主审和审评项目组成员(如医务官和统计师)以及审评部门相应的管理人员合作,在所在的审评处室作出。立项审评应在45 日内完成(包括受理的时间)。此外,立项审评的同时,主审应对该申报资料是否可以进行优先审评判断。 1.2.3 实质审评 PMA立项后,进入实质审评阶段。实质审评中主审负责制订审评时间表、组建项目审评团队、与审评团队中各审评员的沟通、与申办方的各种沟通、组织审评小组内部会议和与申办方的第100天会议等。审评团队一般应包括主审/ 项目组长、医务官、统计师、合规官员、科研工程师、兽医、流行病学家。 1.2.4 专家咨询委员会审评 为了更好地对PMA申请进行评估,FDA的审评员往往需要借助专家咨询委员会的帮助。专家委员会是由大量的非FDA雇员组成,他们大多是各个领域的科学家和临床医生。委员会并不隶属于FDA组织,因此可以提出很多独立性的意见和建议。委员会按照专业性区分,包括心血管仪器委员会、麻醉器械委员会等等,共有16 个。ODE需经过判断是否需要借助委员会的力量来审评,每年召开的专家会数量很少,一般只有在下列三种情况下,才会组织召开专家会:某一类医疗器械首次申请FDA注册时;一些医疗器械同时涉及到的共性问题(比如出现血栓等);对某一类医疗器械进行分类界定时。专家会的全部过程几乎都是对公众公开的。 2.欧盟第三方机构审评模式研究 2.1 组织架构 欧盟医疗器械产品监管模式的特点之一是政府监管部门将产品上市的审批权交由第三方机构执行。欧盟各成员国负责指定第三方机构,即公告机构,并告知欧盟委员会。以SGS(通标标准技术服务有限公司)为例,一个完整的公告机构人员配置及架构应如图4,5。
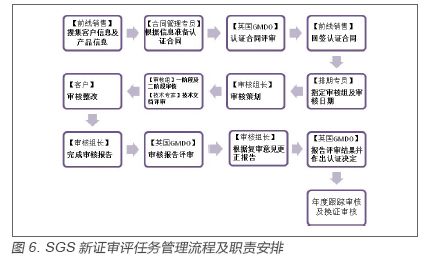
2.2 审评流程 公告机构应具有适当的结构和程序,以确保能够实施与审核过程相关的合格评定和颁发证书。申请人向认证机构的前线客服销售人员提交企业及产品信息,交由合同管理专员准备认证合同并提交给英国公告机构总部(GMDO)进行认证合同评审,前线客服销售依据评审结果与客户签订认证协议。协议签定后,排期专员指定审核组组长,成员及日期。审核组长进行审核策划,并与审核组成员一起完成审核,向申请企业提供不符合项及审核报告。申请企业对不符合项进行整改并回复审核组。审核组收到回复后完成审核报告并提交给GMDO进行报告评审并作出认证决定。业务支持人员依据认证决定制作及发放证书给获证企业。 以SGS为例,首次合格评定和颁发证书的程序及相关人员职责安排应如图6。
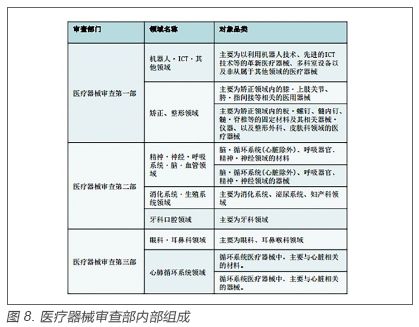
3.日本PMDA 审评模式研究 3.1 组织架构 PMDA是独立行政法人药品医疗器械综合机构。日本依据医疗器械风险高低将医疗器械上市前管理分为三种模式。第一种一般医疗器械风险等级为一级,不需上市前审评,仅向PMDA提交备案即可;第二种管理类医疗器械分险等级为二级,可以由第三方有资质的认证机构审评;第三种高度管理类医疗器械风险等级为三级和四级,由PMDA审查(见图7)。PMDA下设三类医疗器械审查部和体外诊断药审部,此外还有独立的临床评价部、生物学评价部等(见图8)。
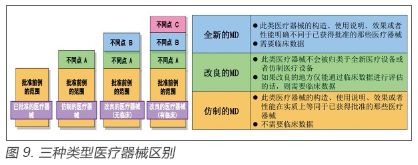
3.2 审评流程 PMDA审评将医疗器械分为全新医疗器械、改良医疗器械和仿制医疗器械,不同类型申报和审评要求不一致,其中仅全新医疗器械产品采取项目审评方式。全新医疗器械、改良医疗器械和仿制医疗器械的判定和区别见图9。
全新医疗器械经受理后,依据产品类别转至相应的医疗器械审查部,由部长指派审查组负责人即主审,由主审组建审查团队,审查团队共同对全新医疗器械资料进行审评。 4.各国项目管理的特点和可借鉴之处 从美国和日本项目审评的类型来看,均适合比较复杂的品类,并不是所有申请均需要,这是优化资源、节省时间的一种考虑。欧盟第三方机构因为本身审评的产品风险较低固无项目管理的内容。所以项目审评是有针对性的,其中日本的全新、改良和仿制医疗器械的分类方式科学合理。 美国和日本的项目分配,均由医疗器械所在科室的部长来分配。这是由于机构框架决定的,因为科室的划分已经将归属的医疗器械品类确定了。而欧盟第三方由专人负责指派审查员进行,这是由于欧盟第三方机构的人员管理模式与其他国家不一样,他是对审评人员具备哪些器械品类审评能力进行确定和评估,属于单个人员管理而非部门管理。所以,从这点上,美国和日本的分配方式更适合我国。 FDA和欧盟第三方均要求主审人员即项目负责人在接收到任务后对项目进行策划,制定时间表。因此,项目策划是在项目管理中重点要借鉴的地方。 审评项目的质量控制,各个国家都有严格的质量控制,尤其对审评时限的要求,质量控制均作为整个体系控制的一部分,有单独的质量部门负责。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号