由于美国较早立法管理医疗器械,美国食品药品管理局(FDA)创立的分类管理办法已被全球普遍接受,因此美国监管医疗器械的法规和模式在国际上有很大影响力和借鉴价值。本版将陆续刊登国家食品药品监管总局信息中心美国医疗器械监管研究报告,系统介绍美国医疗器械上市前、上市后的监管流程,以及美国国家医疗器械评估体系的运转情况,供读者学习借鉴。 1976年5月28日,美国国会通过了《联邦食品药品化妆品安全法》(1938年)的《医疗器械修正案》,确立了医疗器械的监管类别。规定由美国食品药品管理局(FDA)器械与辐射健康中心(CDRH)负责医疗器械生产、包装、标签以及进口企业,还负责监管辐射电子产品(包括医用及非医用),如X射线系统等。1976年医疗器械项目启动时,FDA从事医疗器械工作的人员有180人,到目前已形成一支1700人的队伍,包括技术、临床、工程、法律等专业人员。 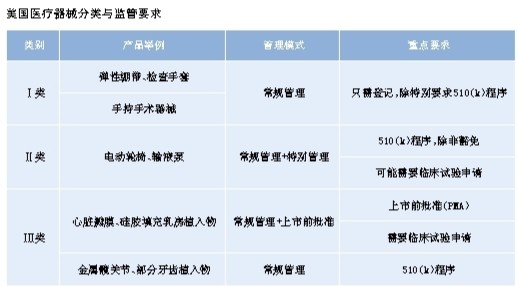 分类方法 美国医疗器械监管思路是基于风险的监管,FDA将医疗器械产品分为Ⅰ类、Ⅱ类和Ⅲ类,监管程度从Ⅰ类~Ⅲ类逐步增强。 Ⅰ类医疗器械 低风险,通过常规管理足以确保其安全性和有效性的医疗器械。对此类器械,FDA首先必须确定有充分的信息支持产品的分类决定。大多数Ⅰ类医疗器械上市前可以豁免通过510(k)(某些Ⅰ类及大部分Ⅱ类医疗器械的上市程序,以联邦法律中的相应章节命名)程序批准。 Ⅱ类医疗器械 中度风险,通过常规管理不足以提供安全性和有效性的合理保证。为减少风险,需要为患者提供信息或特别管理,才能确保其安全性和有效性。特别管理通常包括特别标签要求、强制性绩效标准、上市后警戒等。大多数Ⅱ类器械需要通过上市前通知510(k)。 Ⅲ类医疗器械 高风险,常规管理和特别管理不足以确保产品的安全性和有效性,此类器械作为支持或维持人的生命,或对预防人体伤害特别重要,或有潜在、不明原因疾病或损伤风险,需要通过上市前批准,提供合理的安全性和有效性保证。大多数Ⅲ类医疗器械需要通过上市前批准,有的产品可能通过510(k)程序批准。《医疗器械修正案》通过后的10年里,80%的Ⅲ类医疗器械通过510(k)申请进入市场。 1997年的《美国食品药品现代化法案》及2012年的《食品药品安全与创新法案》,明确了一种针对低到中度风险的新器械产品分类认定方法,建立起此类产品尽早上市的绿色通道,这就是De Novo分类。通过De Novo认定后,产品可以按照Ⅰ类或Ⅱ类医疗器械监管。 管理模式 常规管理 常规管理是1976年《医疗器械修正案》基本规定,适用于所有三类医疗器械,监管内容包括产品注册与登记、上市前通知、修理、换货、记录与报告等9方面。很多监管思路与我国类似。 特别管理 针对Ⅱ类医疗器械,特别管理通常与器械相对应,包括性能标准、上市后警戒、患者登记、特别标签要求、上市前数据要求以及指导原则。 上市前申请 Ⅲ类医疗器械需要上市前审批。 值得关注的是,美国通过立法鼓励医疗器械产品的创新,注重对罕见病群体的人道主义关怀。1990年,美国国会通过《安全医疗器械法案》,创建了针对影响少数人群(8000人/年)的医疗器械监管项目,即人道医疗器械项目,为罕见病人群使用的、临床研究数据难获得的产品上市提供便利。 来源:总局信息中心美国医疗器械监管研究报告 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 










