摘要:近年来,中国的医疗法规不断的完善,基本医疗制度框架已基本搭建完成。IVD产品的注册流程随着管理规范的变化而不断改变,各种注册难点也相继出现。如何避开法律的雷区,并根据法规的指引找到有前景的投资方向,是制定投资决策的首要关注因素。 国家在IVD行业的法律法规已经逐步落地,了解行业政策将带您规避雷区,使得投资更精准、更高效。下面是政策专家、致众科技董事长曾建辉先生在泰山汇举办的“新政下IVD行业投资机遇与新趋势”高端沙龙上所做的关于IVD行业相关政策的部分发言: 一丶中国医疗法规的发展沿革
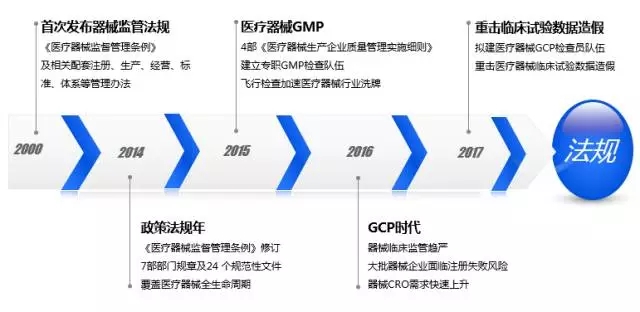
1.萌芽期。中国第一部有关医疗器械的法规是1996年出台的“医疗器械管理办法”。2000年出台了第一部“监督管理条例”,并出台了与之配套的一系列经营、生产、注册、体系的管理办法。当时,产品的核批通过较为宽松,临床证据的取得也相对简单。 2.摸索期。2007年—2014年期间出台的条例被反复修改。其中多次讨论的“注册管理办法”里就加注了“除创新医疗器械以外,我们用以检测和临床注册的产品必须在符合医疗器械生产质量管理体系的环境下生产出来的产品。”这句话意味着大部分企业必须先建厂,待企业的IVD产品拿到注册证后,才能取得生产许可证。 3.成熟期。2014年中国首次提出了医疗器械生命周期的监管体系,从研发到最后生产、临床、注册、销售、售后,都有法规覆盖,尤其这些年,不良反应事件的检测国家也非常重视,各个省中心都设置了专职的机构,叫不良反应中心。在法规的修订方面,国家投入巨大。 4.落地期。2015年至今法规已逐步落地。飞检、临床核查是常规的监管手段。一旦企业进入国家局的核查名单,企业产品的注册难度将会加大。IVD本身就是重灾区,因为做IVD的临床检查速度快,所以监管IVD产品不需要任何机构,三天的时间就能完成。并以拍照、留记录的形式保留临床证据。
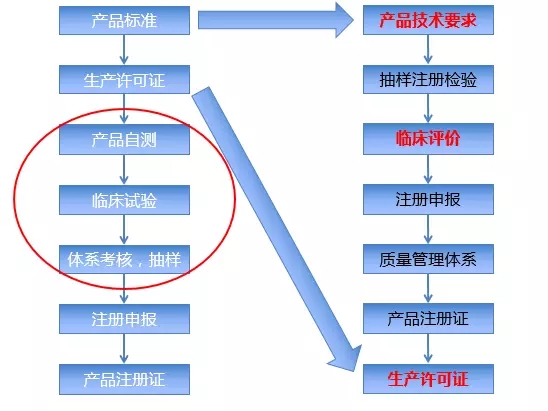
二丶中国IVD产品的注册难点 1相关研究文献可信度较差。中国的很多文献可信度比较差,与临床有效率相比存在较大的差异。很多IVD产品没有注重数据的完整性、真实性和可追溯性。当企业的产品跟相关产品对照的结果不符合法规、结果的时候,缺乏有力的研究证据。 2.新注册办法使流程更复杂。诊断试剂是具有盈利性的,根据2016版的“注册管理办法”,患者参加临床试验时需具有知情权,当医院让受试者参加临床试验时,如果医院让受试者签知情同意书,受试者在很大可能上不会参加试验。这种情况下,试验所需时间和费用会成倍增加。以前做二类产品需要2000元,如今流程复杂化,在很多医院做一个试验项目需要的费用不低于5万元。 3.GCP资质认证提升试验费用。部分试验医生只有拿到GCP资格证才能做项目的研究者,由于拥有GCP资格证的医生只是少数,间接地提升了临床试验费用,与有限的项目经费不匹配,最终导至现在很多医院明确不接IVD的项目。
三丶给投资人的建议 1.提前对企业做合规性的审查。针对近三五年拿到注册证的产品,必须查清对应的数据合规不合规,执照是否已被吊销。在体系分解过程中,把一些不合格的地方罗列一下。未来的IVD行业,竞争成本将更加有序,可以在一个层次上竞争。分解也会进一步加强,无菌IVD会全面覆盖,国家文件也如是反映。 2. 技术创新要遵守监管体系。相关法规是由部分专家制定的,但法规是所有医疗器械相关方,包括医院、企业、监管部门、投资人等都要遵守的。所以,医疗器械的技术创新要遵守整个监管法规体系,如果企业的研发没有遵守国家监管的法规体系,那所有的研发投入最终都难以转化为成果,都是浪费。 3.LDT和CMO将更有前景。从2011年到现在,中国医疗器械注册量持续攀升。IVD创新产品申报类别集中在PCR类和测序类,这类产品注册价格高、注册周期长是普遍性的特点,所以LDT(临床实验室自建项目)一定是未来的趋势之一。3月底国务院已经发文,批准上海自贸区做CMO试点,将来CMO注册检测将会变得快捷,所以CMO也是未来的趋势之一。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号