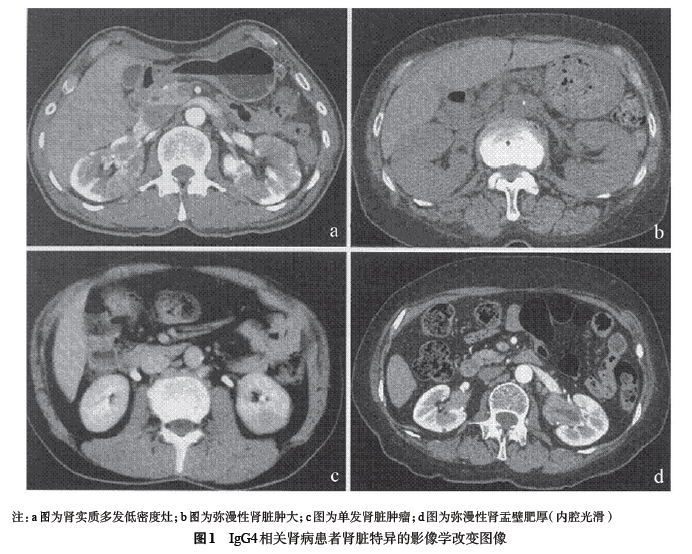
2001 年日本学者滨野等报道了血清IgG4 浓度升高可作为诊断自身免疫性胰腺炎(autoimmune pancreatitis, AIP)的标志物之一,并且发现了AIP患者的胰腺及腹膜后组织中多数有IgG4 阳性浆细胞浸润。 此后,又有学者先后报道了AIP患者的胰腺周围组织、胆管、胆囊、肝脏的门静脉区域、胃黏膜、肠黏膜、唾液腺、淋巴结、骨髓等胰腺外的组织器官中也有较多的IgG4 阳性浆细胞浸润。 此类疾病是以血清IgG4 升高及组织中IgG4 阳性浆细胞浸润为共同表现特征的全身性疾病,因此命名为IgG4相关疾病(IgG4-related disease, IgG4-RD)。 2004 年在世界范围内陆续有文献报告其可合并肾脏损害,主要表现肾脏及其周围器官组织病变,称之为IgG4 相关肾病(IgG4-related kidney disease,IgG4-RKD),肾损害的主要特征是肾小管间质肾炎(tubulointerstitial nephritis,TIN),故又有学者称之为IgG4 相关肾小管间质性肾炎(IgG4-related tubulointerstitial nephritis,IgG4- TIN)。 此后,又相继报道了该疾病影像学上可表现为肾盂、输尿管的肥厚及肿瘤样病变,部分患者可伴有肾小球病变,也可合并慢性硬化性泪腺炎、唾液腺炎而无AIP,少数患者病变也可仅局限于肾脏病变,如果没有对此疾病的全面认识,临床上极易漏诊和误诊,如误诊为肾脏肿瘤等,我国对此病的认识相对较晚,直到2011 年才开始有病例报道。 2011年美国学者Riassian等统计分析了35例IgG4-RKD患者,对照组175 例;日本学者总结分析了41 例IgG4-RKD患者,对照组9 例;先后制订出IgG4-RKD诊断标准。两个诊断标准敏感性均为100%,特异性分别为92.0%和95.1%,内容主要包括:肾脏损伤、血清学、影像学、组织病理学等指标,为临床诊断此病提供了方向。 但由于两个诊断标准依据的病例数均较少,因此,还需在临床工作中进一步验证。该病糖皮质激素治疗多数有效,预后一般较好,因此早期诊断对患者有重要意义。发病机制目前尚不完全清楚,可能主要与过敏或自身免疫功能紊乱有关。 一、临床表现 男性患者多见,占所报告病例数的73% ~86%,患病年龄60 岁以上患者较多,肾脏损伤主要表现尿液检查异常,表现为少量至中等量蛋白尿,偶有大量蛋白尿,尿β2 微球蛋白升高,偶有镜下血尿,肾功能损害比较常见。 日本报告的41 例IgG4-RKD患者血清肌酐平均值约为149.6 μmol/L(1.7 mg/dl)肾功不全患病率为58.5%。美国报告的35例患者血肌肝平均值约为318.2 μmol/L(3.6 mg/dl),肾功能不全患病率为77.0%。 患者发病特点与AIP类似,多数伴有肾外脏器损害,累及器官组织包括胰腺、肝、胆管、胃肠道、唾液腺、泪腺、肺、眼眶、乳房、腹膜后、大动脉、淋巴结、皮肤、垂体、前列腺等,平均受累的脏器为3.4 个;其中最易受累的脏器是唾液腺,其次分别为:淋巴结、胰腺、泪腺、肺等,有最多累及8 个脏器的病例。 也有少数患者仅表现单个脏器损害,如日本报告的41 例患者中有2例患者仅有肾损害(占4.9%),美国报告的35例患者中有6 例仅有肾损害(占17.1%)。 二、血清学检查 多数患者有高IgG 血症、高IgG4 血症和低补体血症,日本报告病例分别占90.2%、100%和53.7%;美国报道病例分别占73.0%、92.0%和56.0%。 外周血嗜酸细胞可升高,日本报道病例78.8%患者有高IgE血症,据此,有学者推测该病的发病可能与过敏有关;美国病例没有报道相关数据。此外,也有报道患者血清抗核抗体和类风湿因子也可升高。 三、影像学特点 美国报道的病例78.0%有影像学异常,日本报道病例也占70.7%。影像学检查表现为双肾多发性低密度损害(呈类圆形、楔形,可累及肾被膜)最多见,其次分别为双侧弥漫性肾脏肿大、单发肾脏肿瘤、弥漫性肾盂壁肥厚(内腔光滑)等。 部分患者表现为肾脏肿块,类似肿瘤样病变,可突出于肾脏轮廓外(见图1)。因此,要注意与肾脏肿瘤进行鉴别。美国报道的35 例患者中有9 例(占25.7%)是因影像学检查异常而首诊,从而进行肾活检或肾切除组织病理检查而确诊。
四、肾脏损害病理特点 主要表现为TIN,但要与其他原因所致的TIN鉴别,其主要特点是:光镜下有弥漫或片状淋巴细胞、浆细胞浸润,病变部位与非病变部位界线清楚,病变不仅限于肾皮质,也常累及肾髓质,甚至肾被膜、腹膜等,而且 IgG4 和IgG阳性浆细胞比例≥ 40%、或IgG4 阳性浆细胞数量> 10 个/HP。 浸润细胞周围有特征性的纤维化,典型呈螺旋环状,又称“鸟眼”征,伴肾小管萎缩(见图2)。电镜下80%以上病变部位肾小管基底膜,可见电子致密物沉积,甚至包曼氏囊、肾间质亦可有免疫复合物沉积。
直接免疫荧光检查主要表现为IgG、C3、κ、λ 沿毛细血管壁呈颗粒样沉积,偶可见沉积于系膜区,呈颗粒样。Raissian 等将IgG4-TIN 肾脏病理分三种类型:(1)急性间质性肾炎,伴少量纤维化;(2)广泛间质纤维化伴炎细胞浸润;(3)寡细胞性纤维化,由此可推断患病时间的长短,并在2 例因肿块切除的肾脏组织中发现临近肿块中心部位纤维化显著而细胞成分较少,肿块周边表现为炎细胞浸润为特征的改变。 可伴有肾小球损害,日本报道的41 例患者中有11 例(占39.3%)伴有肾小球病变,最常见的是膜性肾病(3 例),与其他学者报道一致,其次分别是紫癜性肾炎(2 例),IgA肾病(2 例),毛细血管内增生性肾炎(2 例),膜增殖性肾炎(1 例),系膜增生性肾炎(1 例)。美国报告的35 例患者没有相关肾小球损害的报道。
五、美国肾脏病学会和日本肾脏病学会两个诊断标准的比较 美国肾脏病变学会提出的诊断标准(见表1)中,在细胞浸润最集中区域IgG4+浆细胞> 10个/HP是必备的条件,伴有其他影像学、实验室检查或其他脏器受累中至少1 条即可诊断。
美国纳入的对照组病例数较多(共计175 例),包括82 例回顾2007年肾活检显示有明显的肾小管间质肾炎改变患者,93 例2007 年以外的肾小管间质性肾炎患者,临床表现、影像学和(或)组织学与IgG4-TIN相似。 结果显示,除了IgG4-RKD外,约有8.4%的TIN患者有中等程度的IgG4+浆细胞浸润(11 ~ 30 个/HP),实际上,最近的研究也发现,病理组织上有IgG4 阳性细胞浸润的疾病也不仅限于IgG4-RD,亦可见于其他疾病,如Castlemam病、小血管炎、特发性间质性肾炎、甚或糖尿病肾病等,而此类疾病亦可导致肾损害,有可能造成误诊,需注意鉴别。
 日本肾脏病学会提出的诊断标准(见表2)包括了:确定诊断、拟诊和疑诊条件。从2004 年至2011 年日本肾脏病学会在5 家医院共收集到确诊患者41 例。 纳入对照组的患者9 例,包括3 例嗜酸性肉芽肿性血管炎;2 例肾功不全伴IgG4-RD,但肾活检明确为其他原因导致肾损害;1 例TIN合并干燥综合征;1 例微小病变肾病;1 例低补体血症伴过敏;1 例复发性多发软骨炎。 以血清IgG4 水平升高作为诊断的必备条件。而实际上AIP作为IgG4-RD的代表,亦有报道8% ~ 23%的病例可有血清IgG4水平正常。2010 年Shoji 等报道1 例IgG4 相关炎性假瘤患者,其血清IgG4 水平正常,故该诊断标准也可能会造成一定数量的患者漏诊。 在AIP 诊断标准中包括了糖皮质激素治疗是否有效,尽管IgG4-TIN对激素治疗有效,但其他类型TIN对激素治疗同样也有效,因此,激素治疗是否有效不应作为IgG4-TIN的诊断条件之一,但激素治疗对肾脏肿块或其他脏器受累的疗效反应欠佳或将有益于鉴别诊断。 六、治疗和预后 AIP 首选糖皮质激素治疗,多数病例使用糖皮质激素治疗后有效,肾功能和影像学异常得到显著改善,但有较高的复发率。 美国报告的病例治疗前血肌肝平均为309.4 μmol/L(3.5 mg/dl),治疗后降至了150.3 μmol/L(1.7 mg/dl),随访观察6个月,总有效率为90.5%,但激素减量后有复发的可能性,美国报道35 例患者中19 人应用激素治疗17 人有效,但其中2 人在激素减量过程中复发。 病理组织类型与治疗反应无显著相关性,甚至肾间质广泛纤维化的患者对激素治疗亦可能有效。日本报告的41 例患者中有38 例使用了激素治疗,35 例反应良好,仅3 例肾功持续恶化(1 例患者进入到维持性血液透析,2 例血肌酐升高)。 在Saeki 等研究中,23 例患者中19 例应用了泼尼松治疗,剂量为10 ~60 mg/d,18 例用药4 周后肾功能、补体水平、影像学异常等明显改善,Raissian 等的研究结果也与此类似。 综上所述,就目前文献报道,大部分IgG4-RKD病例糖皮质激素治疗有效,延误治疗将增大肾衰竭的风险,但对于激素治疗的剂量、疗程,以及是否联合应用免疫抑制剂治疗,目前,尚无统一治疗方案,有待于临床进一步研究探讨。 七、发病机制 人类IgG分子根据铰链区结构不同分为IgG1,IgG2,IgG3,IgG4 亚型,IgG4 与其他亚型比较不同之处在于:(1)等电点不同,即在生理中性环境下带负电荷,而其他IgG亚型均带正电荷;(2)在血清中水平最低,占IgG总量的3% ~ 6%;(3)激活补体的能力较弱,与其铰链区较短而活动性差有关。 尽管人们逐渐认识了IgG4-RKD,但对其发病机制尚不完全清楚。对IgG4-RKD发病机制的研究目前多见于AIP。因IgG4-RKD常伴有自身免疫性疾病,血清学检查多种自身抗体阳性,且对激素治疗敏感,因此有学者认为其发病可能与自身免疫功能异常相关。 IgG4-RKD研究目前在日本最活跃,各国家也已经重视,病因及发病机制也正在积极探讨中。已有的研究成果表明,发病主要与自身免疫功能紊乱及过敏有关,如与Th2 及抑制性T 细胞应答体系改变有关,期待IgG4-RKD发病机制研究的进展。 总之,IgG4-RKD最主要特征是特异性的TIN,自2004 年有病例报告以来,已经得到各国肾脏病学者的关注,2011 年以来我国大陆地区已有病例报告。 2011 年美国肾脏病学会及日本肾脏病学会先后制订了IgG4-RKD诊断标准,为临床诊断提供了参照方向,但由于依据的病例数不多,因此还需在临床中进一步验证。随着对IgG4-RKD认识的提高,企望能尽快制定出一个国际统一的诊断标准,治疗方案也有待于进一步规范化。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号