国家重磅发布新版GCP!检验需要重点关注的5个问题
2026-6-10 10:09|
发布者: 沙糖桔|
查看: 333|
评论: 0|来源: 检验智医365网
摘要: 检验科要守住三条线:样本合规、结果可靠、数据可追溯。
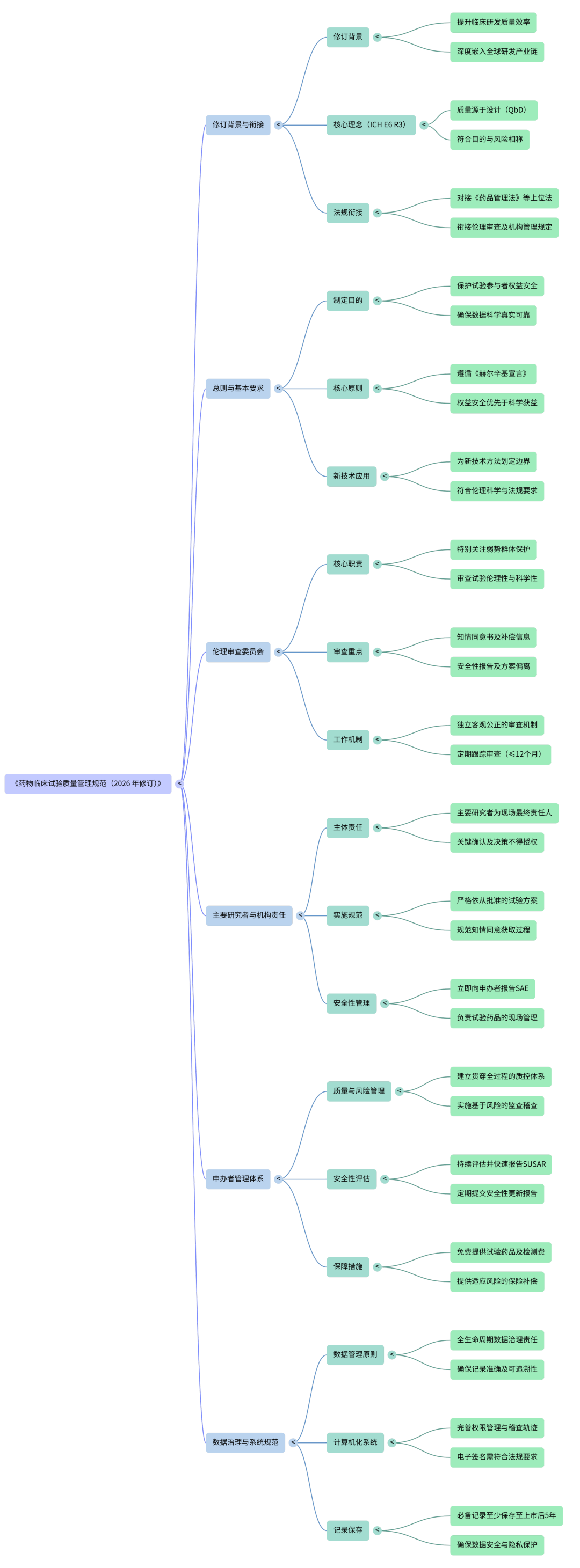
近日,国家药监局、国家卫生健康委、国家中医药局、国家疾控局发布《药物临床试验质量管理规范(2026年修订)》。该规范将自2026年9月1日起施行,2020版GCP同时废止。在药物临床试验中,检验科往往承担血液、生化、免疫、凝血、尿液、微生物、分子检测等项目。有些医院还承担中心实验室、样本处理、样本暂存、生物样本外送、特殊检测配合等工作。也就是说,检验科虽然不是每个项目的“主角”,但很可能是临床试验数据链条中的关键环节。新版GCP对检验科最重要的提醒是:临床试验样本不能简单等同于普通门急诊样本。它既是医疗检测样本,也是临床试验数据来源。样本怎么采、怎么收、怎么测、怎么传、怎么保存、怎么追溯,都会影响试验结果的真实性和可靠性。新版GCP强调,药物临床试验全过程应当遵循伦理、科学和质量标准,目标是保护试验参与者权益和安全,确保数据和结果科学、真实、可靠。临床试验中的检验结果,可能用于入排标准判断、安全性评价、疗效终点分析、不良事件判断,甚至影响试验药物是否继续使用。如果检验结果不准确,影响的不只是单个患者的诊疗,还可能影响整个临床试验结论。临床试验不是只看最终报告。标本采集时间、接收时间、离心条件、保存温度、检测人员、仪器批号、试剂批号、校准质控、复检记录、异常值处理、报告修改记录,都可能成为核查重点。新版GCP新增“数据治理”章节,说明监管关注点已经从“有没有结果”进一步转向“数据从哪里来、谁录入、谁修改、为什么修改、能不能追溯”。这对检验科LIS、HIS、EDC接口、外送检测数据、电子签名、权限管理等提出了更高要求。新版GCP明确,承担生物样品检测分析的实验室应符合相关规定并具备资质,申办者要监督生物样品采集、管理、检测分析、运输、储存和销毁等全流程质量管理。同时,禁止实施与伦理审查委员会批准的试验方案无关的生物样品检测,例如未经批准的基因检测等。临床试验样本不是“多抽了一管血,就可以顺便做点别的检测”。试验方案、知情同意书、伦理批准文件里没有写清楚的检测,原则上不能随意开展。新版GCP要求临床试验数据应从源记录中获得,并保证可靠性和可追溯性。源记录应具有可归因性、易读性、同时性、原始性、准确性和完整性。源记录修改应当可追溯,不能掩盖原始记录,并记录修改理由。对检验科而言,检验报告只是结果呈现,背后还有大量源数据。这些内容在平时可能被视为“后台资料”,但在临床试验核查中,可能就是判断数据是否真实可靠的依据。新版GCP要求,主要研究者应按照试验方案要求和时限,向申办者报告安全性评价所需的不良事件、异常检查结果。检验科不是安全性报告的最终责任部门,但检验科往往是异常信号的第一发现环节。比如肝肾功能异常、血常规明显异常、凝血异常、电解质紊乱、感染指标异常等,都可能触发方案规定的安全性评价、不良事件判断或停药标准。不能只满足于“报告已经发出”,还要关注是否符合试验方案规定的时限和路径。新版GCP的数据治理要求非常明确:临床试验数据应包含元数据,包括稽查轨迹;计算机化系统要满足数据可靠性、可追溯性和安全性要求;系统应具备用户管理、权限管理和稽查轨迹;电子签名应符合我国电子签名相关要求。这意味着,检验科信息系统不能只追求“能出报告”,还要能回答几个问题:对参与临床试验较多的医院,检验科应联合信息科、GCP机构办公室,对LIS、HIS、EDC、外送检测平台之间的数据流进行梳理。尤其要关注手工录入、Excel中转、截图传输、纸质报告补录等高风险环节。新版GCP强调,药物临床试验机构开展临床试验,应建立质量管理体系,并确保有效运行。主要研究者和临床试验机构如授权个人或单位承担临床试验相关职责,应建立完整工作制度,实施有效监督管理,确保其具备相应资质并能正确履职。只要检验科参与临床试验检测,就应当纳入项目启动、培训、授权、SOP、质控、偏差处理和文件归档体系。- 项目启动前,确认试验方案、检测项目、样本类型、采样要求、报告时限;
- 样本接收时,核对受试者编号、采集时间、样本状态和方案要求;
- 检测过程中,保留仪器、试剂、质控、复检和异常处理记录;
- 报告发布后,确保数据传输、报告修改、危急值通知可追溯;
这次新版GCP不是要求检验科重新建立一套“复杂体系”,而是提醒科室把已有的质量管理体系与临床试验要求接上。建议先做三项工作。第一,梳理本科室正在参与或曾经参与的药物临床试验项目,明确哪些项目由检验科承担检测任务,哪些项目涉及样本暂存、处理或外送。第二,检查临床试验样本与普通样本是否有区分管理机制,特别是采集要求、接收标准、拒收标准、报告时限、异常值反馈、样本保存和销毁记录。第三,联合GCP机构办公室和信息科,梳理临床试验检验数据从LIS到研究病历、EDC或申办方系统的流转路径,重点排查手工录入、人工转抄、报告修改、权限管理和稽查轨迹问题。新版GCP对检验科的影响,不在于增加多少新表格,而在于重新明确了一个基本逻辑:临床试验中的检验结果,不只是“一张报告”,而是药物研发证据链的一部分。检验科要守住三条线:样本合规、结果可靠、数据可追溯。只有这样,才能真正支撑临床试验质量,也能在检查、核查和稽查中经得起追问。国家药品监督管理局、国家卫生健康委、国家中医药局、国家疾控局:《关于发布药物临床试验质量管理规范的公告(2026年第50号)》《〈药物临床试验质量管理规范(2026年修订)〉问答文件》
|
声明:
1、凡本网注明“来源:小桔灯网”的所有作品,均为本网合法拥有版权或有权使用的作品,转载需联系授权。
2、凡本网注明“来源:XXX(非小桔灯网)”的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。其版权归原作者所有,如有侵权请联系删除。
3、所有再转载者需自行获得原作者授权并注明来源。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号