过去一年FDA发布了哪些重要声音?
2024-1-11 16:57|
发布者: 沙糖桔|
查看: 2276|
评论: 0|来源: 霍普金斯MedTech欧美资讯
摘要: 新冠EUA时代结束,开启510(k)新时代
作者Ethan 弹指间2023年已然成为过去式,FDA在一年中发表了不少政策,也举办过若干次会议。作为最大的甲方,FDA的每次动向都对美国整个IVD行业产生着深远的影响,对于许多国内心心念念想要出海的企业来说有着重要的参考价值。作为新年开门的第一篇法规相关推送,笔者精心整理了去年最值得注意的两件大事。新冠EUA时代结束,开启510(k)新时代
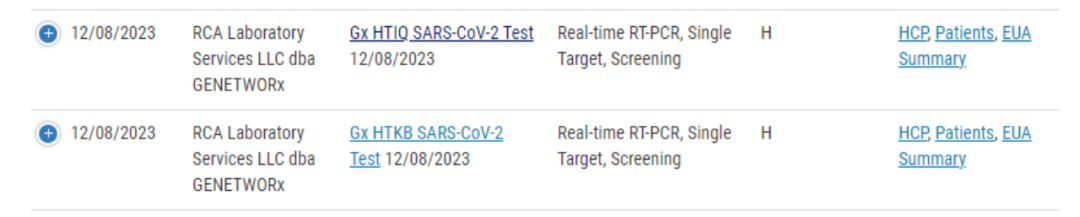
美国拜登政府在去年的5月11日正式结束了紧急公共卫生状态(Public Health Emergency,PHE)。与此同时,FDA也在不断收紧PHE时期发布的政策,年初连续发布了两篇关于医疗器械和紧急使用授权(Emergency Use Authorization,EUA)产品的过渡期指南,让各家厂商做好准备。于是,所有PHE相关政策在2023年11月7日之后(即自新冠PHE结束180天后)全部失效。但是值得注意的是,EUA并不会因为PHE时期的政策失效而失效。也就是说,目前新冠EUA仍然存在且生效。这是因为,PHE隶属于《公共卫生服务(PHS)法》第319条,而EUA源于《联邦食品,药品和化妆品(FD&C)法案》第564条EUA并不依赖于PHE声明,只有当美国卫生部秘书主动终止EUA时,EUA才能正式结束。因此,我们现在仍然能看到FDA新批的EUA证书。最新批准的是12月8日RCA Laboratory Services公司的Gx HTIQ SARS-CoV-2 Test和Gx HTKB SARS-CoV-2 Test。同时,由于美国新冠疫情逐渐回归为常规流行病流行程度,FDA对新冠检测产品的态度也从EUA转向了De Novo和510(k)。De Novo是新型中风险医疗器械首次申请美国上市的渠道,此前FDA从未批准过同类产品。而510(k)是中风险器械根据此前批准过的器械进行实质性同效证明从而上市。EUA毕竟是特殊时期的特殊上市渠道,谁也不能确定EUA什么时候会突然终止,因此FDA鼓励厂商通过传统上市渠道(De Novo和510(k))进行上市,来保障美国居民能够随时随刻获得新冠检测试剂,这是根本所在。目前,在传统上市渠道中,FDA一共批准了14个新冠分子测试,1个新冠抗原测试和2个新冠血清测试。其中,一共3个新冠分子POC产品,不知道大家发现了没,目前FDA批准的新冠传统上市产品里唯独缺少了新冠抗原OTC产品。但往往新冠抗原OTC产品是最重要的产品类型,为什么我们这么说呢? 后疫情时代的新冠需求
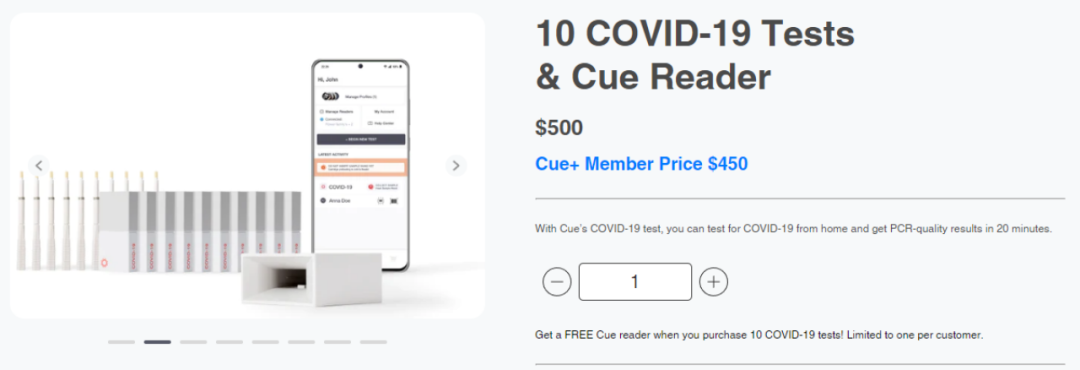
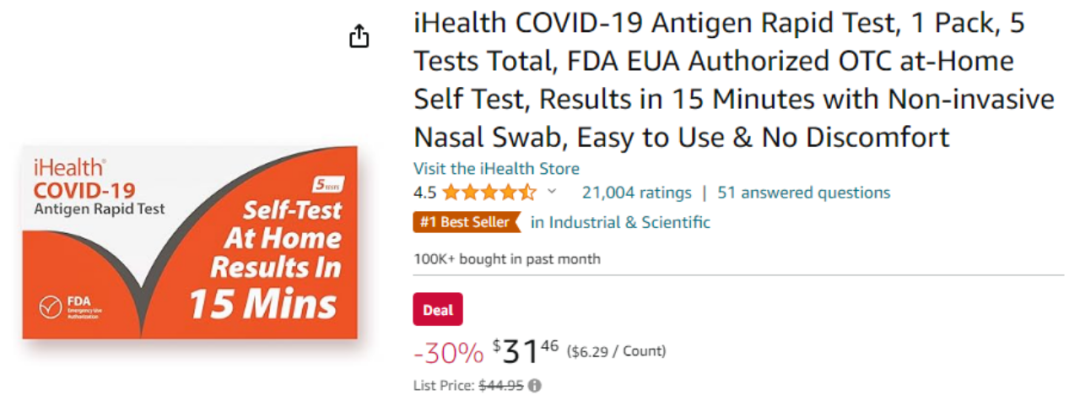
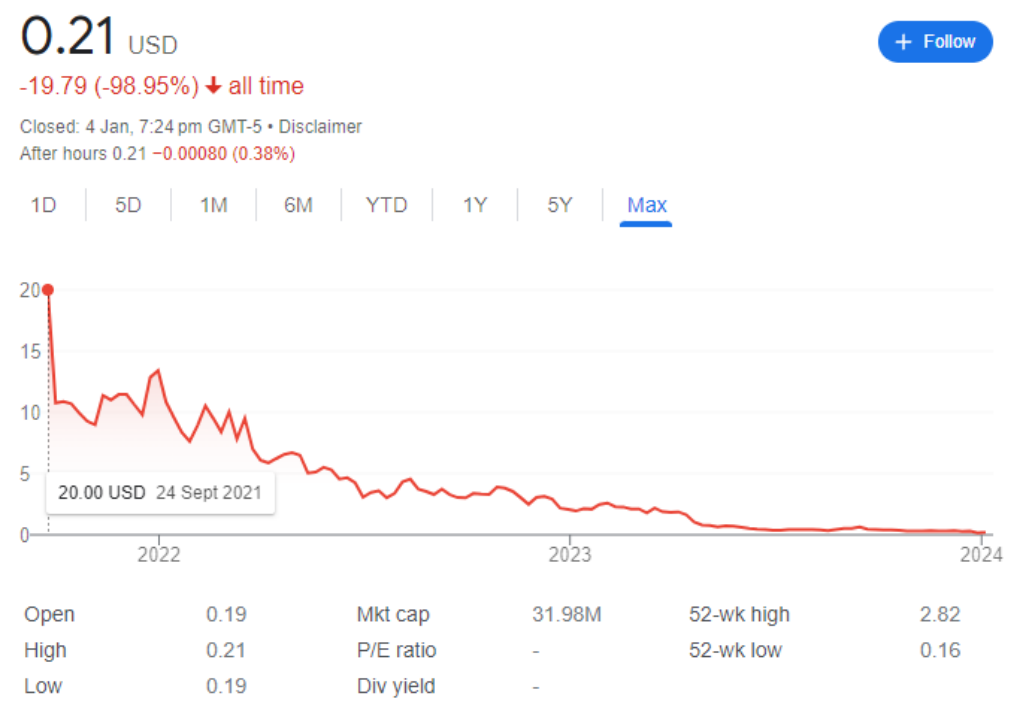
大家都有过疫情期间需要自己捅鼻子、做新冠抗原检测的经历,在家做检测可比自己到核酸亭、医院做方便多了。因此,基本上所有人都会同意抗原OTC是新冠赛道最主流的产品,其他种类只能做到补充的作用。可能会有人会问,同样是居家检测,为什么分子OTC就做不到?看看价格和股价就知道了。Cue Health的新冠检测是FDA当下批准的唯一分子OTC产品,但是需要配套它的仪器(Cue Reader)一起使用。初期需要花费500美金购买10个test,附赠一台Cue Reader。之后每10个test需要另外支付500美金。这样换算下来,每个人每次测试test需要50美金。接下来看看新冠抗原OTC,目前亚马逊上卖的最火的新冠产品就是九安医疗的抗原OTC测试,一人份检测售价6.29美金。上个月在亚马逊平台上直接卖出了10万多份,这里还没有算上九安官方平台上的销量。6.29美金对比50美金,直接8倍的大差价。在二者性能没有太多区别的情况下,绝大多数人还是会捂住钱包,购买九安的新冠产品。两者销量的反差也直接反映在了二者的市值上。Cue Health股价从刚开始的20美金直接跌到现在的0.21美金,当前市值3200万美金;而九安医疗股价从最低的5元一路飙升至现在的44.7元,当前市值217.3亿元人民币。如果Cue Health的产品线或战略方向不作出及时的转变,恐怕公司股价只会一直跌下去,直到最后的退市。因此,未来新冠抗原OTC一定是后疫情时代的主流需求!FDA拟监管LDT
要说哪件事情能跟新冠510(k)一样重磅,那莫过于去年FDA计划监管LDT了。FDA在9月31日提出了拟议LDT的政策,4年5步走,计划在政策正式生效后的4年内逐步要求LDT与其他IVD产品一样遵循IVD需遵循的一切法规,从遵循医疗器械报告、质量管理体系要求,再到最后的递交上市申请。FDA的意思很明确,就是未来美国市场上基本上只有IVD产品。 | | | | | 结束与医疗器械报告 (MDR) 要求以及更正和删除报告要求相关的一般执法自由裁量权 | | | 结束MDR、更正和删除报告、QS以及上市前审查要求以外的其他要求的一般执行自由裁量权 | | | | | 发布正式政策3.5年后,但不得早于2027年10月1日 | 结束高风险IVD产品上市前审查要求的一般执法自由裁量权 | | | 结束中低风险IVD产品上市前审查要求的一般执法自由裁量权 |
10月31日,FDA举行了LDT网络研讨会,在会议中FDA表达了和政策里一致的监管LDT的态度,对于拟议政策进行了更多的详细阐释。未来实验室不再能使用仅供研究使用(Research Use Only, RUO)组件,因为这些组件需要遵循IVD的合规性和质量管理要求。实验室也不再能修改部分组件来让某个获批产品更符合自己实验室的流程,并提高效率了。修改后的产品同样需要递交上市前申请。此次LDT政策不可谓不严,并且FDA希望赶在今年美国总统大选前,4月份通过该政策,以防新国会可能推翻LDT。基本上所有美国人都认为,该政策正式通过后,将会给实验室行业带来毁灭性的打击。总体而言,一共有三方面的原因:第一个方面,像Labcorp和Quest一样大的实验室联盟因为本身拥有足够的资源和财力,LDT转传统上市渠道对于它们来说并不困难;但对于小型实验室来说就是一场灾难,本身就要准备美国CMS和所在州卫生部的定期审查,还要抽出物力财力来申请产品上市几乎是不可能的事情。最后结果只会变成马太效应:强者愈强,弱者愈弱;第二个,实验室的创新技术也会被LDT新政策所压制,因为新技术想要上市的话基本需要走De Novo和PMA渠道,这两个渠道相对于510(k)来说更加困难,对于小实验室来说就更加天方夜谭。第三个,目前监管实验室的权利归CMS所有,FDA并不拥有管辖权。FDA就算要管,还要跟CMS算清楚账,划分好各自权力范围。总结来说,FDA提出的LDT政策与美国卫生部本身鼓励创新、保障美国人民随时都拥有最新最好的医疗技术的原则基本背道而驰。这也是为什么美国实验室行业内基本上呈现一边倒的反对趋势,基本上知名的美国医疗协会都提出了抗议。再加上像Advamed这样的大型医疗游说团体也在持反对态度,FDA面临的阻力可想而知。在HMC看来,FDA监管LDT的法规愿景是好的,但基本不可能被国会通过。
|
声明:
1、凡本网注明“来源:小桔灯网”的所有作品,均为本网合法拥有版权或有权使用的作品,转载需联系授权。
2、凡本网注明“来源:XXX(非小桔灯网)”的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。其版权归原作者所有,如有侵权请联系删除。
3、所有再转载者需自行获得原作者授权并注明来源。


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号