快速、准确和全面的诊断是确定疫情并改进病原体监测以防止进一步传播的关键。近年来,高通量下一代测序技术(NGS)创造了不需要培养或先验知识就能全面分析样品的可能性,这种方法被称为宏基因组测序(mNGS)。然而,目前的短读NGS技术需要专门的实验室、熟练的人员和高额投资,不能提供实时信息,只提供DNA片段信息,限制了随后的分类或功能分析。 最近,牛津纳米孔技术公司(ONT)开发的便携式、实时、长读测序技术为快速、全面的现场微生物诊断和监测铺平了道路。尽管纳米孔测序本身可以在小型设备上进行,但目前大多数DNA提取、文库制备和数据分析方法仍然需要非便携式设备和稳定的互联网和电网接入,这限制了潜在的使用。近年来,已经开发了几种设备和方法来实现现场宏基因组数据的生成。例子包括Bento Bio, SuperFastPrep-2和Claremont Bio OmnilyseX和DNAexpress试剂盒。此外,笔记本电脑供电的ONT VolTRAX允许在便携式设备上自动建库。 近日,一组来自比利时的研究团队在杂志Scientific reports上发表了一篇题为“Development of a portable on-site applicable metagenomic data generation workflow for enhanced pathogen and antimicrobial resistance surveillance”的文章。在本研究中,作者将重点放在湿实验室方面,结合Claremont Bio OmnilyseX, Bento Bio Pro和VolTRAX设备开发了一种便携式现场适用的mNGS方法。为了优化数据生成方法,作者使用鸡粪样本作为测试案例,因为家禽肠道微生物组已被证明含有多种抗生素耐药基因ARGs。作者逐步改进并最终建立了一个湿实验室流程,可产生高质量的纳米孔mNGS数据。并使用spike-in的模拟群落,作者将分类和抗性组分析性能与其他测序工作流程进行了比较。
图片来源:Scientific reports 主要内容 优化一种快速宏基因组DNA提取方法 为了减少设备需求和样品处理时间,选择Claremont Bio DNAexpress试剂盒来开发现场DNA提取工作流程。该方法包括在含有微珠和裂解缓冲液的电池驱动的Omnilyse X bead-beating管(B)中均质和裂解样品,然后是基于DNAexpress (D)柱的DNA纯化方案(如下图)。对方案进行了各种修改(缩写为BD)。然而,BD产生的DNA纯度较低,260 nm/230 nm吸光度比(A260/230)约为0.80,且经各种尝试后无法解决。因此,作者开发了BQ方法,保留了电池供电的Omnilyse X bead-beating(B)步骤,但用Quick-DNA HMW磁珠(Q)试剂盒取代了DNA柱纯化过程(如下图),发现这将A260/230比率提高到1.91,并将DNA产量提高了约500 ng。方案BQ的最终版本包括使用Omnilyse X管以1.5 V进行bead-beating,然后进行Q纯化和一轮AMPure XP纯化,bead/样品比为0.4(如下图)。它返回的高纯度DNA数量与目前基于实验室的EQ方法相当(酶(E)裂解然后是方法Q纯化)。
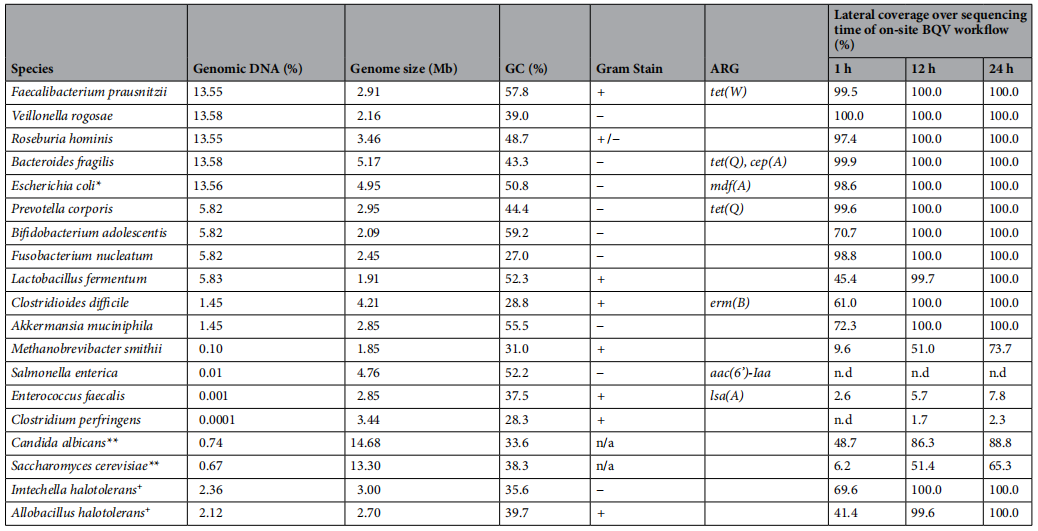
宏基因组工作流程概述。图片来源:Scientific reports 不同宏基因组工作流程的表现评估 使用加标模拟群落比较测序工作流程 为了评估各种宏基因组DNA提取方案的性能,将鸡粪样本加入由ZymoBIOMICS肠道微生物组标准(GMS)组成的DMC,并结合ZymoBIOMICS spike-in Control I,其中包含两种不存在于粪便背景中的海洋物种。此外,作者报告了通过ResFinder检测到的DMC参考基因组中的ARG含量,以及在添加DMC的鸡粪便样本中最终现场mNGS工作流程BQV产生的每个基因组随时间的横向覆盖率(测序reads覆盖的参考基因组比例)(如下图)。
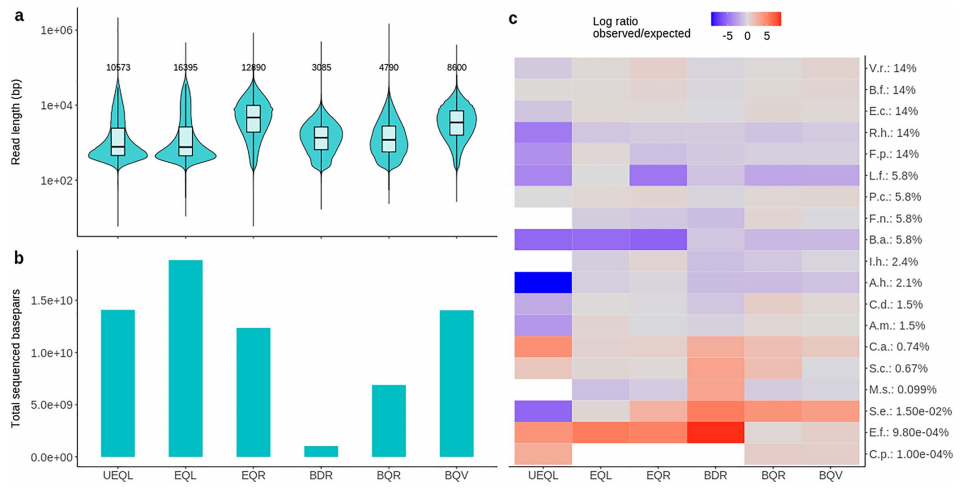
模拟群落(DMC)的组成和特征,以及横向基因组覆盖率。图片来源:Scientific reports 流程对测序通量和读长的影响 为了比较现场DNA提取方案与其他工作流程的性能,作者生成了六个纳米孔测序文库(表3)。 UEQL:Unspiked-enzymatic lysis-quick-DNA magbead HMW-ligation sequencing; EQL:enzymatic lysis-quick-DNA magbead HMW-ligation sequencing, EQR:enzymatic lysis-quick-DNA magbead HMW-rapid sequencing, BDR:bead-beating-DNAexpress-rapid sequencing, BQR:bead-beating-quick-DNA HMW magbead-rapid sequencing, BQV:bead-beating-quick-DNA HMW magbead-voltrax sequencing. 作者比较了各种方案的测序输出和统计数据(如下表)。总的来说,酶解文库(UEQL, EQL和EQR)在测序通量方面优于bead-beading文库(BDR, BQR), 但BQV除外。酶解文库也表现出更高的读长N50。然而,bead-beading的执行速度要快1 - 2.5小时。与BDR相比,BQR和BQV中bead-beading强度的降低改善了DNA片段大小,从而获得更高的读取长度和通量。最后,BQV中较高的输入DNA量,以及VolTRAX文库制备中额外的自动磁珠清理,进一步增加了读取长度和通量,导至与基于实验室的文库EQR相当的通量,尽管读长N50仍然较低。
测序工作流程概述。图片来源:Scientific reports 测序工作流程影响分类学分类 作者将每种方法生成的reads与含有DMC参考基因组的数据库进行了比对,并将每个DMC物种的观测相对丰度(RA)与理论相对丰度(TRA)进行了比较(如下图)。简而言之,酶促法对几乎所有物种都产生了更长的reads,除了L. fermentum,并且在物种之间显示出较大的reads长度分布差异。相比之下,bead-beading方法通常导至较短读长,物种之间的读长分布差异较小。接下来,作者使用了一个广泛的分类数据库来进一步研究所有工作流程中检测到的粪便微生物背景。与未加标背景(UEQL)相比,一些背景物种,如Bifdobacterium gallinarum和乳酸杆菌科的成员在酶解工作流程中代表性不足(EQL, EQR)。对于bead-beading方法(BDR, BQR, BQV),情况并非如此。
每个工作流程的测序统计及DMC检测的比较。 图片来源:Scientific reports 现场工作流程使快速分类鉴定成为可能 给定一个已知大小和丰度的加标基因组,以及每个映射读数的测序时间,就可以确定达到一定基因组覆盖率所需的测序运行时间。作者评估了表现最好的现场工作流程BQV,DMC每个类群随时间的横向覆盖率(1h,12h,24h)。RA > 5%的大多数DMC物种在测序的第一个小时就接近全基因组覆盖,除了b . adolescentis和 L. fermentum,在整个测序数据中代表性不足。这些物种以及丰度在1%到5%之间的物种,在测序12小时后,已经达到完全测序。对于白色念珠菌和酿酒酵母,100%的基因组覆盖率从未达到。结果显示,BQV流程在唯一鉴定物种的数量方面优于其他流程,但无法验证是否真阳性。 利用全长抗性基因进行耐药组学分析 为了分析抗生素耐药基因ARG信息,作者通过将其映射到ResFinder数据库来识别全长ARG。背景UEQL中,ARG多样性最高(图b)。在对加标粪便样本执行的工作流程中,BQV方法的ARG read count最高,其次是EQL、BQR、EQR和BDR方法(图c)。总之,在背景(UEQL)中检测到的大多数ARGs都能被其他工作流程检测到,尽管丰度不同。
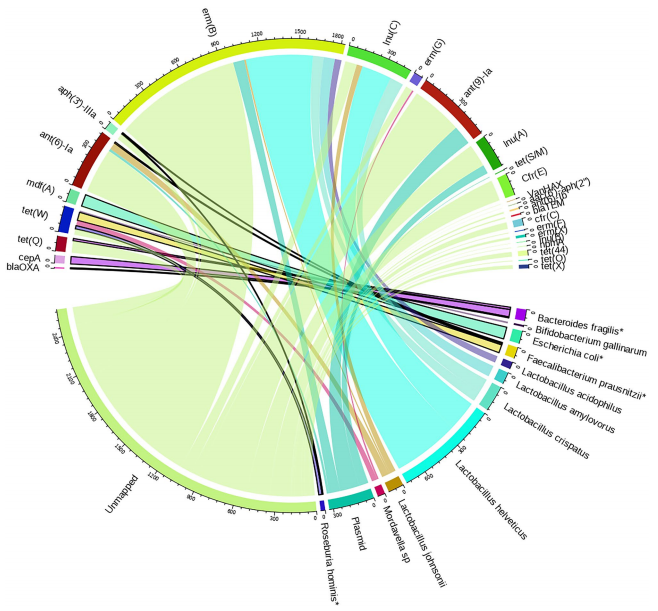
不同工作流程物种和ARG检测的比较。 图片来源:Scientific reports 利用基因组背景将抗性基因与其宿主关联 由于长读可以提供ARGs侧翼区域的额外信息,作者试图将ARGs与其微生物宿主物种关联起来。BQV产生的此类组合最多。在所有实验中,超过一半的全长ARG reads不能关联到任何宿主。粪便样本中大多数鉴定的ARG宿主属于乳酸杆菌科,与氨基糖苷和MLS-B耐药基因有关,其次是具有四环素耐药的Mordavella和Bacteroides属。
BQV方法生成的抗性基因-宿主关联。 图片来源:Scientific reports 总结与讨论 便携式设备上的实时、高通量测序的发展为使用mNGS进行快速、全面和无培养的诊断和监测开辟了可能性。在这项研究中,作者开发了一种适用于纳米孔测序的现场DNA提取和文库制备方法,该方法与目前基于实验室的方案相当或优于后者。作者还演示了使用spike-in的模拟群落来比较工作流程,并发现了分类组成和ARG含量的巨大差异,强调了使用适当的DNA提取和测序方法的重要性。 优化的现场适用方法(BQV)允许在3小时或更短的时间内从复杂样品中生成现场mNGS文库,通过使用便携式设备进行细胞裂解,DNA纯化和文库制备,所得到的文库纯度高,产生的测序通量与当前基于酶解(EQL, EQR)的实验室方法相似,同时绕过耗时的孵育步骤。总体而言,BQV方法检测到了大多数加标的DMC,以及来自背景样本的高ARG和分类多样性。ARGs通常位于可移动的遗传元件中,这些元件通常在分类数据库中缺失。因此,为了提高ARG宿主归属的可靠性,未来的发展应侧重于完善分类数据库和改进分类算法。调整这些计算方法,使其适用于低资源环境,可能需要执行一个完整的样本,以在现场产生工作流。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号