一、消息靠谱吗?
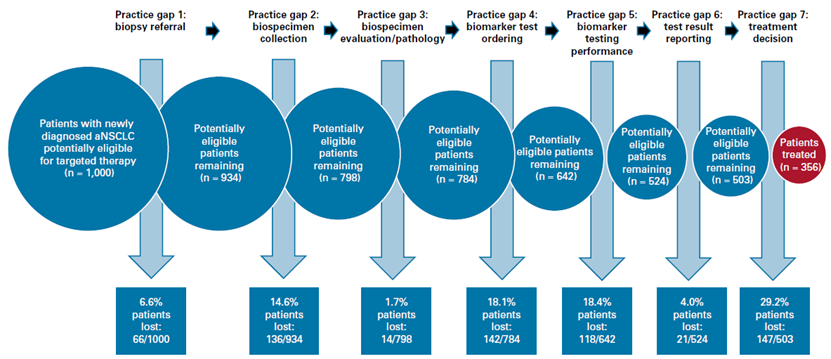
不过,可以看出FDA意识到伴随诊断在使用中带来的阻碍超过了预期的获益(以非小细胞肺癌为例,超过64.4%的病人因为检测反而阻碍了用药,见图1),医生也因为价格/可及性等原因,并没有时刻按照“一药一伴随“的标签开处方。
除了现行伴随诊断使用规则造成的障碍和资源浪费,会上还提到第二个问题:目前的检测没有外部验证(external validation),实际操作中的检测失误很可能被低估。 这两个由Friends提出的问题代表了真实世界的声音,符合以患者为中心的出发点。 CDRH的试点项目仍在研究筹备中。FDA很清楚,知识产权驱动的伴随诊断检测行业将随之发生商业逻辑的变化,但因知识产权造成的资源浪费才是FDA在乎的。 从伴随诊断的发展历程看, FDA的法规也确实是朝着更inclusive的方向演化的(图2)。
二、会怎么操作? “Minimal Performance Criteria”的试点项目如果roll-out,根本目的便是给用药检测松绑,顺便更严格地规范检测在真实世界的可靠性。 因此“minimal performance criteria”的试点项目可能会考虑:
首先,不难看出该提案对LDT的指向性。 临床实际应用中,一个biomarker往往对应多个伴随诊断试剂盒,每个IVD试剂盒都对应指定的仪器平台,独特的判读指南,需要厂家提供培训,从成本、时间和空间的角度,一个biomarker就需要医院/第三方实验室巨大而重复的投入。 其次,从议案、试点再到政策落地、运行,很可能还要经历长时间的窗口和磨合期。 涉及伴随诊断规范的部门并不止Pazdur所在的Oncology Center of Excellence,还有和CDRH 以及Office of Commissioner下属的Office of Combination Products (OCP)。 每五年重新授权一次,赋予FDA对药品注册收费权的Prescription Drug User Fee Act (PDUFA)也对处方药的配套软件等有法规要求。 “minimal performance criteria”必然会受益于LDT政策的落地,而VALID法案至今还没有敲定监管LDT的方案。 三、有哪些问题需要解决?
引领者和追随者会做出不同的选择。
最后 在「伴随诊断:从“卖水”开始绘制复合版图」中提到过,医院/药厂/中心实验室/第三方检测/IVD厂家,都有各自的利益诉求,伴随诊断作为一个法规驱动的市场,应以“为患者服务”为长期目标发展技术创新 —— “为患者服务”包括但不限于:更早识别患者、多biomarker的平台化检测技术、自动化判读流程。 正因为NGS等平台化、高通量诊断技术的创新与迭代,才带来了LDT,甚至“minimal performance criteria”的可能性。技术创新始终是行业良性发展的动力,新提案会是应用上的补充,不是创新的捷径。 -END- References Sadik H., et al. Impact of Clinical Practice Gaps on the Implementation of Personalized Medicine in Advanced Non-Small-Cell Lung Cancer. JCO Precision OncologyNo.6 (2022) Pamela Ebrahimi. (2022 September 26-29) Approval of Companion Diagnostics by the U.S. FDA: Exploring the Current Regulatory Environment & Important Hurdles to Overcome. 12th Annual World CB&CDx Summit, Boston, MA |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号