脓毒症导至全球20%的死亡,仅在美国就造成了20-50%的医院死亡。早期诊断和识别潜在的微生物病原体对于及时和适当的抗生素治疗至关重要,这对脓毒症的生存至关重要。然而,在超过30%的病例中,没有发现致病病原体,这反映了当前基于培养的微生物诊断的局限性。更增加了复杂性的是,需要有效地将脓毒症与非传染性全身性疾病区分开来,这些疾病在入院时临床上通常表现相似。 随着宏基因组下一代测序(mNGS),脓毒症诊断的局限性可能会被克服。血浆无细胞DNA测序的最新进展扩大了宏基因组诊断的范围,实现了对源自不同感染解剖部位的循环病原体核酸的微创检测。然而,血浆DNA宏基因组学的临床影响受到了质疑,因为经常鉴定临床相关性不确定的微生物,无法检测导至肺炎的RNA病毒,以及在排除感染存在方面的效用有限。 全血转录图谱分析通过捕获区分感染性和非感染性以及病毒性和细菌性感染的宿主基因表达特征,有可能减轻这些限制。然而,由于转录谱分析只捕获宿主对感染的反应,它不能提供脓毒症病原体的精确分类鉴定,这限制了该方法在单独使用时的实用性。 近日,一组来自UCSF的研究团队在杂志Nature microbiology上发表了一篇题为“Integrated host-microbe plasma metagenomics for sepsis diagnosis in a prospective cohort of critically ill adults”的文章,在文章中,研究团队研究了一个重症成人的前瞻性队列,以开发一种结合宿主转录谱和mNGS的脓毒症诊断方法。通过将机器学习应用于mNGS数据,研究团队评估了宿主和微生物特征,将微生物导至的脓毒症与非传染性危重症区分开来。最后研究团队还证明血浆核酸可以用于检测宿主和微生物,以进行精确的脓毒症诊断。
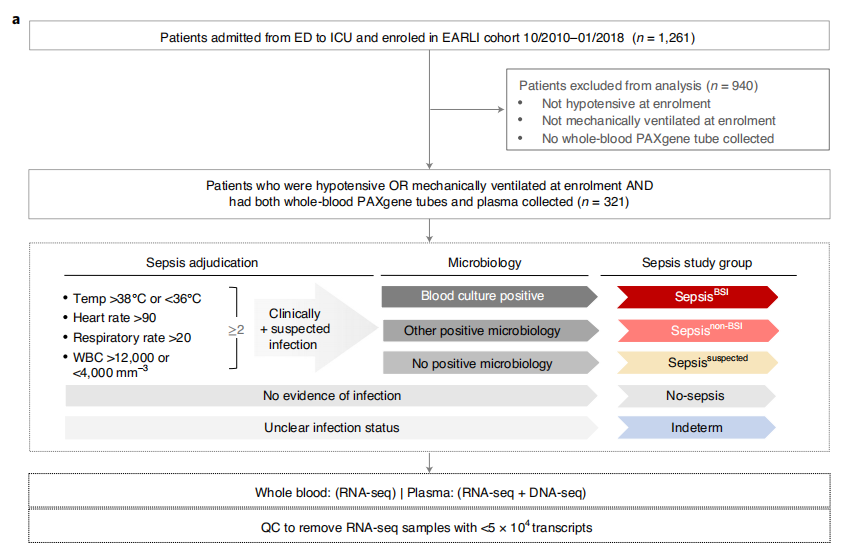
图片来源:Nature microbiology 主要内容 研究队列的临床特征 研究团队对两所医院急诊科(ED)收治至重症监护室(ICU)的重症成年人进行了前瞻性观察研究(下图)。根据脓毒症状态将患者分为五个亚组。这些患者包括:(1)临床判定脓毒症和微生物学确认的细菌血流感染(SepsisBSI),(2)临床判定的脓毒症和经微生物学确认的非血流感染(Sepsisnon-BSI),(3) 临床微生物检测阴性的疑似脓毒症(Sepsissuspected),(4)没有脓毒症证据且对其危重症有明确的替代性解释的患者(No-sepsis)或(5)状态不确定的患者(Indeterm)。除一名患者(无脓毒症组)外,所有患者均表现出≥2全身炎症反应综合征(SIRS)标准。
研究流程图。图片来源:Nature microbiology 脓毒症的宿主全血转录特征 研究团队首先通过对全血标本进行RNA测序(RNA-seq),评估了临床和微生物学证实的脓毒症(SepsisBSI、Sepsisnon-BSI)患者与无感染证据(No-sepsis)患者之间的转录差异(n = 总计221)。识别出P<0.1的差异表达(DE)基因共5807个。基因集富集分析(GSEA)证明了脓毒症患者中性粒细胞脱颗粒和先天免疫信号相关基因的上调,同时与翻译和核糖体RNA处理相关的途径也下调(图b)。
脓毒症的宿主全血转录特征。图片来源:Nature microbiology 用于脓毒症诊断的宿主全血转录组分类器 研究团队基于全血基因表达特征构建了“通用”脓毒症诊断分类器。研究团队采用bSVM学习方法来选择最有效区分脓毒症患者(SepsisBSI和Sepsisnon-BSI)和无脓毒症(No sepsis)患者。bSVM模型在保留的验证集中,AUC为0.82。此外,在10个随机生成的验证集上获得了0.85的AUC(图c)。
宿主全血转录组分类器。图片来源:Nature microbiology 血浆RNA用于脓毒症诊断的宿主转录组分类器 为了测试血浆DNA测序能否提供有意义的基因表达数据,研究团队对与全血样本匹配的血浆样本的RNA进行了测序并进行DE分析,以评估脓毒症患者(SepsisBSI和Sepsisnon-BSI)和无脓毒症患者(No sepsis)的差异基因。值得注意的是,几个顶级差异表达基因之前报道过脓毒症生物标志物(例如,CD177升高,HLA-DRA抑制),这表明血浆RNA具有生物学相关的转录组特征(图d)。 使用相同的bSVM方法产生一个分类器将SepsisBSI和Sepsisnon-BSI患者与非脓毒症患者区分开来,在保留的验证集中,AUC为0.77。在10个随机生成的验证集的AUC为0.90(图e)。
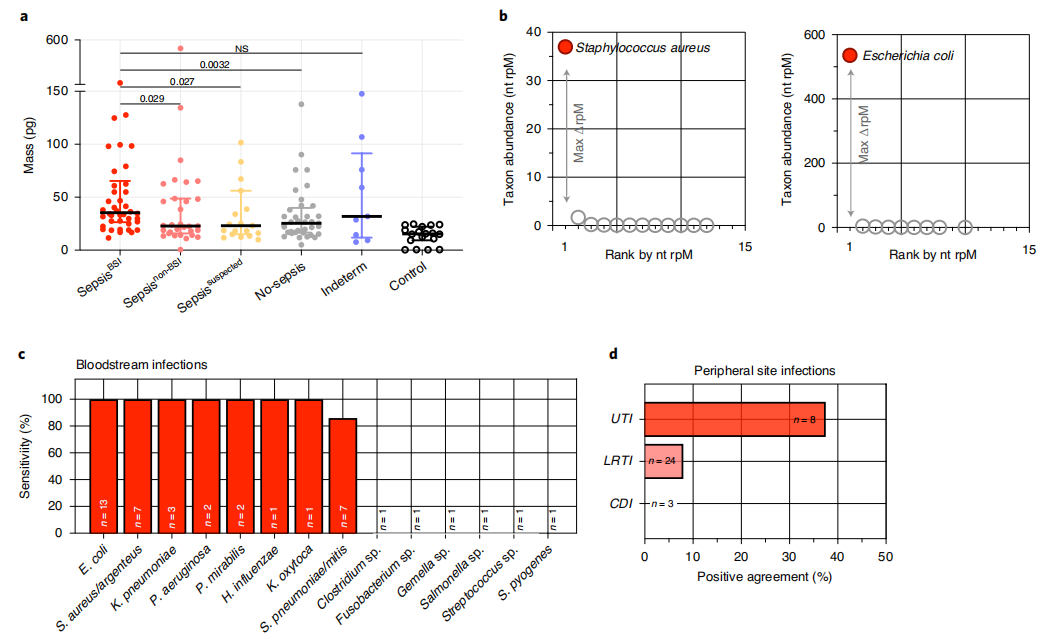
宿主血浆转录组分类器。图片来源:Nature microbiology 从血浆核酸中检测细菌性脓毒症病原体 研究团队开始了微生物宏基因组分析,结果发现,与其他组相比(不确定组除外),SepsisBSI组的微生物质量明显更高,阴性对照水样中的微生物质量显著降低(图a)。接下来,研究团队使用IDseq流程进行细菌病原体检测及分类比对,随后使用先前开发模型(RBM),在mNGS数据中从数量较少的共生或污染微生物中识别出了脓毒症病原体(图b)。 研究团队研究了宏基因组RBM病原体预测与细菌血培养数据的一致性。结果显示宏基因组RBM对血液培养的敏感性为83%,并因病原体而异,范围从0%(例如艰难梭菌)到100%(例如大肠杆菌、金黄色葡萄球菌)。在无脓毒症组中,10/37(27%)患者的被RBM模型识别出病原体,相当于73%的特异性。
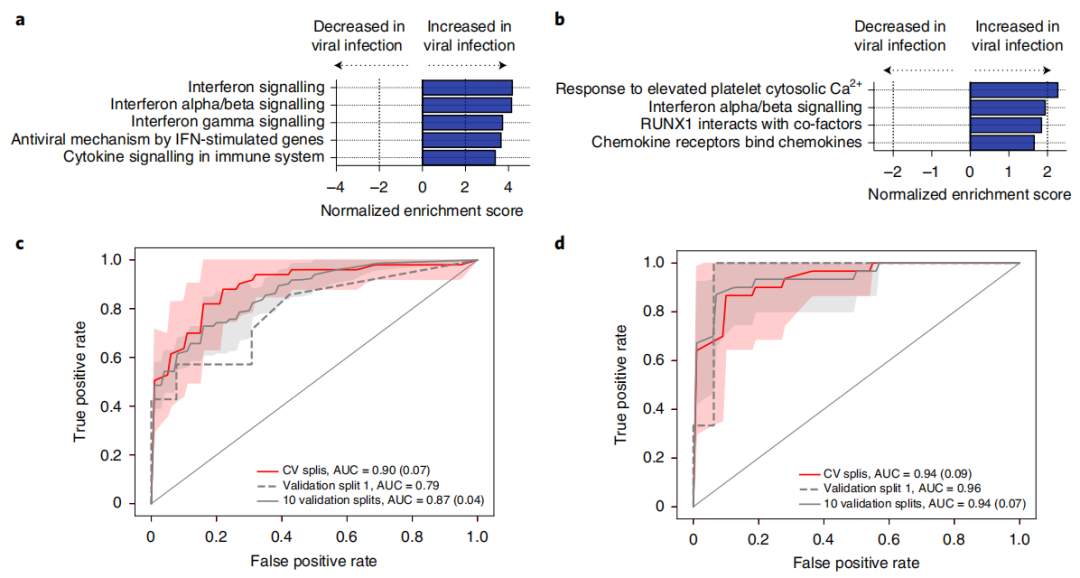
微生物宏基因组识别脓毒症病原体。图片来源:Nature microbiology 利用宿主转录组图谱鉴定病毒感染 13种呼吸道病毒中只有1种(8%)可以通过血浆RNA mNGS检测到。可否使用宿主反应,通过使用全血或血浆转录组学数据,对SepsisBSI和Sepsisnon-BSI组中病毒性脓毒症的患者进行差异基因表达分析来识别病毒性脓毒症?GSEA证明,在来自全血(图a)和血浆(图b)数据中,与细菌性脓毒症患者的样品相比,与干扰素信号相关的途径和抗病毒免疫重要的基因在病毒性脓毒病患者的样品中富集。 研究团队利用这一宿主特征构建了病毒脓毒症的二级bSVM诊断分类器,选择差异表达基因作为潜在的预测因子。在保留的验证集中 ,AUC为0.96。10个随机生成的验证集的AUC为0.94(图d)。
基于宿主基因表达的病毒性脓毒症检测。图片来源:Nature microbiology 基于血浆核酸的宿主微生物脓毒症综合诊断模型 研究团队开发了一个概念验证集成宿主+微生物模型。应用这些规则可以检测到SepsisBSI组42/42例(100%)病例和Sepsisnon-BSI组30/31例(97%)病例,总体敏感性为72/73例(99%)。该概念验证模型在无脓毒症组中的特异性为29/37(78%)。 在19名临床判定为脓毒症但医院微生物检测阴性(疑似脓毒症)的患者中,14名(74%)被归类为脓毒症。对于不确定组,综合宿主+微生物模型将8/9(89%)分类为脓毒症阳性。其中,两个被确定为细菌病原,一个被病毒宿主分类器确定为病毒感染。 总结与讨论 脓毒症被定义为宿主对感染的失调反应,然而现有的诊断只专注于检测病原体或评估感染宿主的特征。在这里,研究团队将宿主转录谱与广泛病原体检测相结合,以在入院时准确诊断危重症患者的脓毒症。此外结果证明,可以对血浆中的循环RNA和DNA进行宿主微生物宏基因组学整合方法,这是一种广泛可用的临床样本类型,在基于宿主的传染病诊断中具有以前未被认可的效用。 此研究有几个优势,包括创新性地将血浆RNA转录组学用于脓毒症诊断,结合宿主和微生物mNGS数据开发脓毒症的诊断方法,详细的临床表型分析,以及一个具有系统性疾病的重症成年人的大型前瞻性队列。它也有一些局限性。首先,如上所述,在不同时间采集的不同样品上进行mNGS和血液培养,因此观察到的与临床微生物检测的一致性可能被低估了。其次,几份血浆样本的宿主转录物不足,无法进行基因表达分析,导至血浆样本量比全血样本量小。这一限制可能在未来的研究中通过增加血浆RNA的输入量来解决。 总之,结合宿主基因表达图谱和血浆核酸的宏基因组病原体检测,可以准确诊断脓毒症。未来的研究需要验证和测试这种与培养无关的诊断方法的临床影响。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号