自2007年以来,全基因组关联研究(GWAS)已经确定了数千种常见SNP与身高之间的关联,主要是通过对欧洲血统参与者的研究。迄今为止发表的最大的成人身高GWAS关注共同变异,报告了712个基因座中多达3290个独立关联,样本量高达700000人。成人身高具有高度遗传性,易于测量,比任何其他人类表型提供了更多的共同遗传关联。此外,大量基因与骨骼生长障碍有关,这些基因在GWAS所绘制的基因座中丰富。这些特征使身高成为评估共同遗传变异在定义多基因人类表型的遗传和生物学结构中的作用的一个有吸引力的模型特征。 近日,由全球数百位研究学者共同在顶刊Nature上发表了一篇题为“A saturated map of common genetic variants associated with human height”的文章。在这篇文章中,研究团队使用5百多万个不同祖先的个体的全基因组关联研究数据,绘制了与成人身高的常见遗传关联图,并评估该图在变异、基因组区域以及可能的致病基因和基因集方面的饱和度。结果表明,这组常见变异在欧洲血统的人群中达到了预测准确性的极限,并在很大程度上饱和了与身高相关的基因组区域和可能相关的基因组;未来的工作将需要将预测准确性扩展到其他祖先的群体,以解释更罕见的遗传变异,并更明确地将相关区域与个别可能的致病基因和变异联系起来。
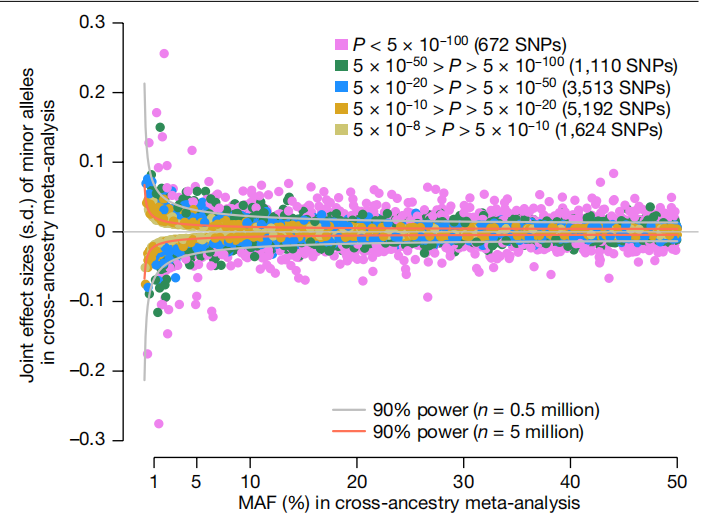
图片来源:Nature 主要内容 Meta分析确定12111个与身高相关的SNP 研究团队对来自GIANT和23andMe的281项研究的5380080名个体进行了遗传分析。样本包括4080687名主要来自欧洲祖先的参与者(占总样本的75.8%);472730名参与者,主要是东亚血统(8.8%);455180名具有典型混血血统的西班牙裔参与者(8.5%);293593名以非洲血统为主的参与者,大多数是非洲裔美国人,具有非洲和欧洲血统的混合血统(5.5%);将这五组参与者或队列分别称为EUR、EAS、HIS、AFR和SAS。 研究团队从多祖先Meta分析中识别准独立GWS SNP,并最终确定了12111个准独立GWS SNP,下图显示了12111个关联中次要等位基因的频率和效应大小之间的关系。
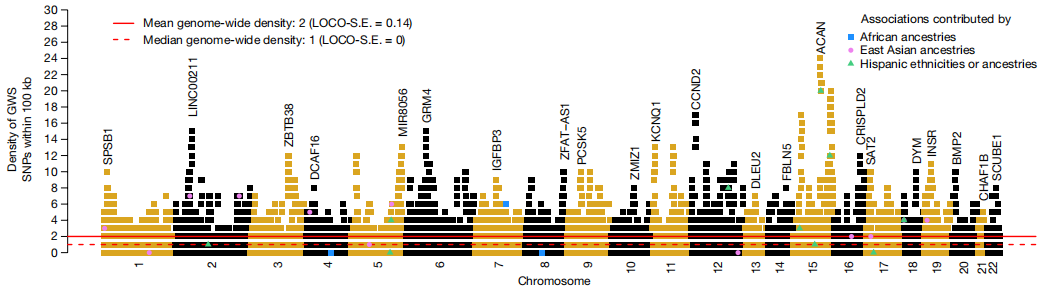
次要等位基因的频率和估计效应大小之间的关系。图片来源:Nature 身高相关SNP的基因组分布 研究团队观察到69%的GWS SNP与另一个相关的、条件独立的GWS单核苷酸多态性共享其位置(下图)。研究团队评估了从OMIM数据库中筛选的462个常染色体基因周围的信号密度,这些基因包含导至骨骼生长异常综合征的致病性突变。结果发现,高密度的身高相关SNP与这些OMIM基因的位置密切相关。因此,这12111个GWS SNP彼此非随机地聚集在一起,靠近已知的骨骼生长基因。 在ACAN附近的第15号染色体上观察到最大密度的条件独立关联,ACAN是一种在矮小和骨骼发育不良综合征中突变的基因,其中25个GWS SNP共同定位在相互100 kb范围以内。
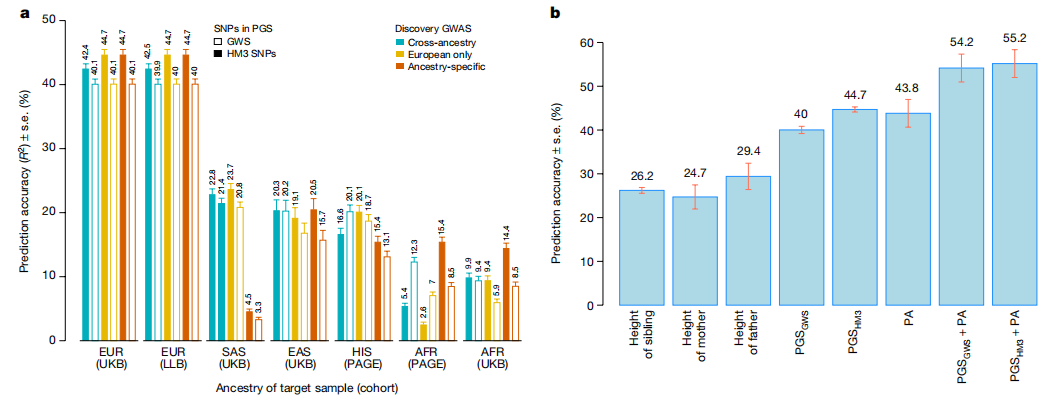
Brisbane plot显示了身高独立遗传关联的基因组密度。图片来源:Nature 样本外预测精度 研究团队根据GWS SNP(以下简称PGSGWS)和所有HM3 SNP(下文简称PGSHM3)量化了身高多基因评分(PGS)的准确性。 总体而言,在大多数祖先群体中,祖先特定的PGSHM3始终优于其相应的PGSGWS。在EUR中,PGSHM3的准确率为44.7%,高于之前发表的基于SNP的身高预测值。总之,对具有欧洲血统的样本进行的方差估计和预测分析表明,12111个GWS SNP几乎解释了所有身高相关的差异,并且将基于SNP的PGS与家族史相结合显著提高了预测准确性。相比之下,估计和预测结果都显示出非欧洲血统样本的明显衰减,与之前的研究一致。
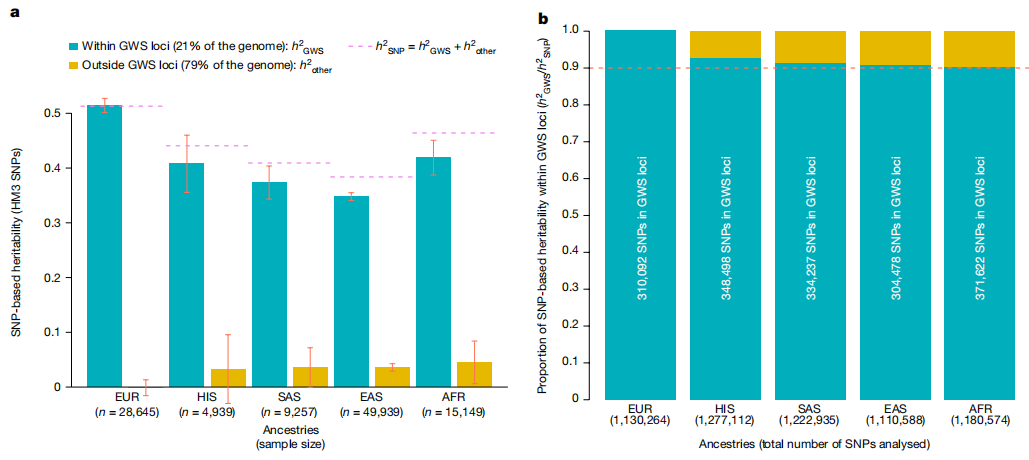
PGS的准确性。图片来源:Nature GWS SNP可解释的身高变异 GWS基因座的累积长度约为647 Mb或基因组的约21%(假设基因组长度约为3039 Mb)。 为了估计GWS SNP所解释的遗传力部分,研究团队设计了方法估计了基于SNP的身高分层遗传力。结果表明,这7209个GWS基因座中的SNP解释了EUR中约100%的身高变异和所有非EUR组中超过90%的身高变异,尽管这些SNP来自基因组的不到21%(下图)。 需要在非欧洲血统的队列中进行进一步的工作,以绘制这些区域内未捕获的剩余5-10%的基于SNP的遗传力。
GWS基因座内HM3 SNP解释的身高变异。图片来源:Nature GWAS发现、样本量和祖先多样性 此大型研究提供了一个机会,可以量化GWAS样本量和祖先多样性的增加对变异、基因和生物途径的发现有多大影响。在变异和基因组区域的水平上,GWS SNP的数量随着样本量的增加而稳定且几乎呈线性增长,当n值大于250万时,新发现基因座数量饱和 ,上升趋势开始减弱。 总之,这些分析表明,增加GWAS样本量不仅可以提高预测准确性,而且可以更清楚地了解变异的基因组分布。 总结与讨论 常见的单核苷酸多态性(SNP)可以共同解释人类身高中40-50%的表型变异,但识别特定变异和相关区域需要巨大的样本量。使用5百多万个不同祖先的个体的全基因组关联研究数据,我们发现,12111个与身高显著相关的独立SNP几乎占据了所有基于SNP的遗传力。这些SNP聚集在7209个非重叠基因组片段内,平均大小约为90 kb,约占基因组的21%。在整个基因组中,独立关联的密度不同,密度增加的区域富含生物学相关基因。在样本外预测中,12111个SNP占欧洲血统人群表型变异的40%,但在其他血统人群中仅占约10-20%。影响大小、相关区域和基因优化在各祖先中相似,这表明预测准确性的降低可能由连锁不平衡和相关区域内等位基因频率的差异来解释。总体而言,这项研究提供了包含绝大多数常见身高相关变异的特定基因组区域的全面地图。尽管这张地图对于欧洲血统的人口来说已经饱和,但需要进一步研究才能在其他血统中达到同样的饱和。 结果已经表明,关联的信号密度不是随机分布在整个基因组中;相反,在以前与孟德尔生长障碍相关的基因周围更可能检测到相关的变异。此外,我们观察到,不同祖先的队列之间存在很强的遗传重叠关联。在具有非欧洲血统的人群中观察到的大多数身高遗传关联与欧洲血统人群中确定的关联非常接近且存在连锁不平衡。 研究有很多局限性。首先,这些SNP仅部分捕获了常见的遗传变异。这一结果强调了身高相关位点的广泛等位基因异质性。另一个局限性是,大约24%的发现样本是非欧洲血统。目前没有具有足够大的多祖先参考面板的分析工具可用于正确解决如何在多祖先研究中识别条件独立的关联。当尝试在包含多个信号的基因座中跨越祖先时,变异的精细定位仍然是一个特别的挑战(对于身高来说通常是如此)。第三个限制是,由于目前此类数据的可用性有限,无法对具有非欧洲祖先的人群的遗传关联进行有力的复制分析。最后,与所有GWAS一样,效应基因的确定以及基因和变异体影响表型的机制仍然是一个关键瓶颈。 身高已被用作研究人类多基因特征(包括常见疾病)的模型特征,因为它具有高遗传性和相对容易测量性,能够实现大样本量。关于遗传结构、额外GWAS发现的样本量要求以及最初对身高进行多基因预测的范围的结论,基本上与常见疾病的结论一致。如果这项研究的结果也可以推断为疾病,这将表明,大幅增加的样本量可以在很大程度上解决归因于有限SNP(和小基因组区域)的共同变异的遗传力。这些变异和区域将涉及基因、调控元件和通路的特定子集,这些基因、调控元素和通路与解决功能、机制和治疗干预问题最为相关。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号