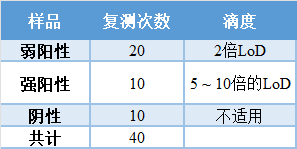
本部分内容摘自《用于分子诊断测试的家庭标本采集设备开发商模板》的“性能评估”部分。 H. 性能评估 在你的家庭采集试剂盒开发过程中,应进行以下建议的验证研究。[对于每项验证研究,你应提供一份研究方案,包括详细的、分步骤的描述,说明如何准备样品和如何进行测试。你还应包括所有验证研究的完整研究线数据,以兼容Excel的电子表格格式提供给所有验证研究。] 1) 家庭采集样品稳定性研究设计: 如果你的试剂盒将使用泡沫或包裹的聚酯鼻拭子在0.9%盐水、PBS或干管中运输,你可以参考Quantigen Biosciences[1]在盖茨基金会和UnitedHealth集团的支持下进行的夏季稳定性研究,不必为样品运输进行自己单独的夏季稳定性研究。 如果您的试剂盒将使用泡沫或包裹的聚酯鼻拭子在干管中运输,您也可以参考Quantigen Biosciences在盖茨基金会和联合健康集团的支持下进行的冬季稳定性研究,而不必为样品运输进行自己的单独冬季稳定性研究。 如果你运送的是干拭子,你也应该提供一个补液SOP供实验室使用,并提供补液方案的验证数据。 建议的研究设计是验证家庭收集的鼻拭子样品的介质(你应该测试所有你打算用来运输的介质),或唾液。测试应包括20个2倍LoD的添加样品(弱阳性)和10个5 ~ 10倍LoD的添加样品(强阳性)。我们还建议测试10个阴性样品以监测假阳性。加注的拭子样品应通过在拭子上加注病毒并将拭子置于培养基中(如果合适)来产生。稀释后的临床样本可用于加注,也可以使用加注了灭活的全病毒的临床样本。理想情况下,加注材料应该用最接近临床样品的临床基质来制备。 表 | 样品组合
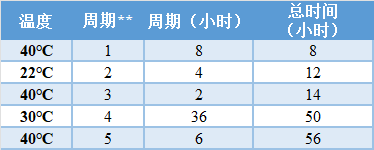
运输研究旨在模拟家庭样品采集和运输过程中的以下情况: Ø 客户运送样品前的样品储存 Ø 样品放在邮箱或投递箱中等待取货 Ø 取样后的运输条件,当样品被运到测试实验室时 一般验收标准: Ø 弱阳性样品:与预期结果的一致性 ≥ 95%。 Ø 强阳性样品。100%与预期结果一致。 Ø 阴性样品。与预期结果100%吻合。 下表描述了每个温度曲线,以复制最坏情况下的运输条件(针对春/夏和冬),在运输前在客户家等待8小时,然后再进行48小时的运输周期。如果您计划允许对通过3 ~ 5天邮件运输的样品进行测试,请扩大下面的运输研究时间。如果您计划在收到样品后在实验室现场储存一段时间,或在装运前在采集现场储存,这也应作为额外的周期添加到稳定性研究中。下表中的完整样本应通过以下的温度和时间进行循环,然后用将与你的采集试剂盒一起使用的SARS-CoV-2分子诊断测试进行测试,产生循环阈值(Ct)。FDA希望样品不仅保持阳性,而且Ct值不会明显增加(超过3Ct)。 表 | 夏季概况*
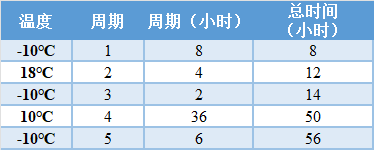
**循环周期指的是连续事件(例如,在不同温度之间循环,持续时间(小时))。 表 | 冬季概况*
*周期2至5的运输条件是按照国际安全运输协会(ISTA)2007年的运输标准(48小时的国内货运),其中周期3和5的温度从35℃提高到40℃。循环周期1(8小时)包括收集样品和运送样品之间的时间延迟。剩余的时间(48小时)涵盖了美国大陆范围内的国内运输。循环期是连续的,每个循环期所需的【周期(小时)】在表中列出。每个周期期结束后,【总时间(小时)】按周期期的小时数递增。 2) 可用性研究: FDA 建议在启动临床协议研究之前完成可用性研究(就本模板而言,可用性研究是评估预期的非专业用户在预期使用条件下遵循标签说明收集、包装和运输样品而不出现严重使用错误的能力),以解决可能影响临床协议评估的说明、包装和运输程序的潜在问题。在计划这些研究时,你可以联系FDA以获得关于你的收集和运输程序以及快速参考说明(QRI)的反馈。如果在临床协议研究之前进行,从这个评估中发现的任何使用错误可以用来在开始临床研究之前修改你的指示和/或程序,然后你将验证最终的指示在临床表现研究期间使用。 FDA建议采用以下研究设计来评估家庭收集和邮寄样品到CLIA认证的实验室进行测试的说明的可用性: Ø 测试应包括至少30名参与者,并在实际使用环境或模拟环境中进行。如果你计划将儿童纳入你的预期使用人群,你的研究应包括每个预期年龄组至少15名儿童。
Ø 整个工作流程应由使用该试剂盒的每个参与者执行,包括试剂盒注册、样品收集、样品包装,以及将预先准备好的标签邮寄给实验室。 Ø 采集时应只使用快速参考说明。我们鼓励开发补充性的在线指导材料/视频,但在研究期间不应使用。 Ø 在样品采集过程中应观察参与者(亲自或通过远程视觉监控,如视频会议),并注意所有的困难。 Ø 在整个过程完成后,应向用户发放调查问卷,以表明试剂盒和样品采集的易用性,以及了解如果步骤执行不正确的后果。如有需要,参与者应能提供意见。 Ø 实验室人员应在交货时按照加入程序检查包装和样品,并注意所有的包装错误和样品的可接受性,以便进行测试。 Ø 参与者应包括代表不同教育水平和年龄的人。有过医学或实验室培训的参与者应被排除在外。理想情况下,应排除以前有自我采集经验的参与者。如果你很难招收天真的用户,你可以招收一些有自我收集经验的用户,但是这个信息应该被记录在你的数据中。 Ø 研究期间收集的样本应使用人类样本控制检测法进行样本充分性测试。这可以包括使用EUA授权的包括人类样本对照(如RNaseP)的检测方法来确定用户是否采集了足够的样本。或者,实验室可以对每个样品进行单独的FDA批准的RT-PCR测试,以人类内务基因为目标。FDA愿意考虑评估足够样本存在和/或质量的替代方法。 Ø 该研究应该有预先确定的接受标准和确定的策略,以减轻研究中发现的错误的风险(例如,修改说明)。 为减少观察到的错误而进行的重大修改可能需要额外的可用性数据,以确保这些修改能有效地减少错误的发生。 关于进行可用性研究的更多信息,请参阅FDA指导文件Applying Human Factors and Usability Engineering to Medical Devices[2]。 [请向FDA提供在可用性研究中使用的说明版本,以及你的可用性研究中产生的总结和行数据。] 3) 用户理解力: 用户理解力研究的目的是客观地评估目标用户对标签中关键元素和概念的理解/理解。FDA建议进行一项研究,以验证和确认用户对检测结果(例如,阳性、无效和阴性结果)和使用说明的理解。[你应该提供数据来验证用户在快速的时间范围内(例如,24 ~ 48小时)获取他们的检测结果。] 如果你不进行这项研究,最好的做法是让医护人员(HCP)联系有阳性和无效结果的病人(电话或短信)。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号