据药智数据梳理,2021年新药注册申请共1933件,同比2020年增长68.09%;其中申请临床1756件,申请上市177件。且值得提及的2021年,国产新药首次IND品种数量超过600个;首次获批的国产新药数量达到了23个。中国正在向着国际创新药研发的中坚力量发展。 根据美国国立卫生研究院的数据显示,我国已经是世界上开展临床试验数量第二多的国家。然而,新药研发从药物发现到申报上市是一个充满冒险与挑战且周期漫长的过程;其中IND申请是新药研发生命周期至关重要的一环。 那么该如何提高IND申报通过率?什么情况下需要提交 IND 申请,又需要提前准备些什么资料?是创新药企加快新药进入临床研究阶段仍亟需思考与探讨的问题。今日,笔者就带领大家一起学习IND注册流程及资料要求,助力药物研发。 IND(Investigational new drug),一般是指尚未经过上市审批,正在进行各阶段临床试验的新药。IND申请,即新药研究申请,目的在于向药监部门提供数据证明药物具备开展临床试验的安全性和合理性,获准后方可开展临床试验。 IND注册流程 新药研究上市主流程
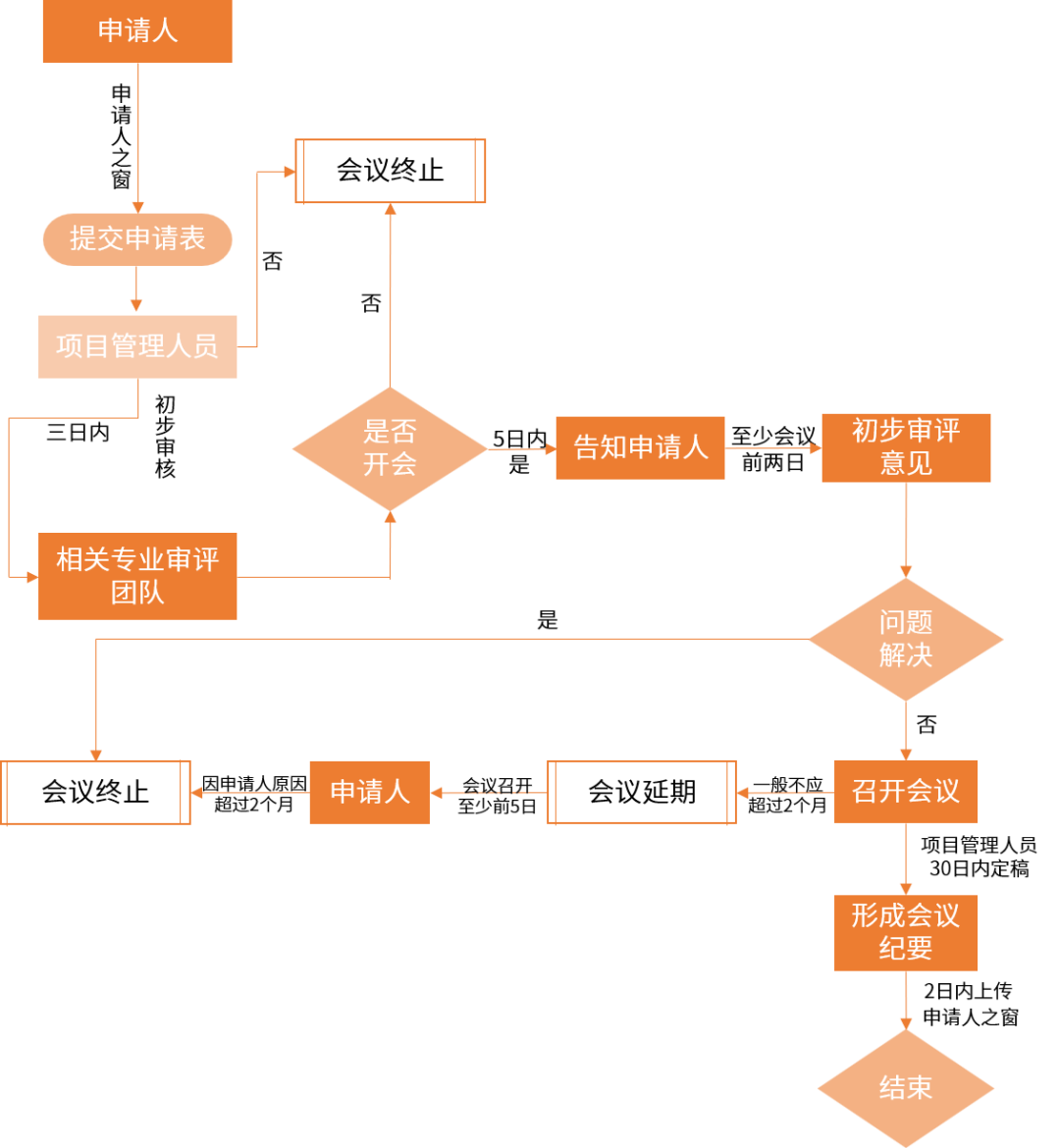
Pre-IND会议 根据国家药监局药审中心发布的关于《药物研发与技术审评沟通交流管理办法》的通告(2020年第48号)及国家药品监督管理局发布的《关于调整药物临床试验审评审批程序的公告》(2018年第50号),沟通交流会在药品上市过程中具有重要意义。 沟通交流,系指在药物研发过程中,经申请人提出,由药审中心项目管理人员与申请人指定的药品注册专员共同商议,并经药审中心适应症团队同意,就现行药物研发与评价指南不能涵盖的关键技术等问题所进行的沟通交流。适用于创新药物、改良型新药、生物类似药、复杂仿制药以及一致性评价品种等研发过程和注册申请中的沟通交流。会议最终形成的共识可作为研发和评价的重要依据。 01沟通交流会会议分类: 沟通交流会分为I类、II类、III类三种会议类型,申请人可在临床研发不同阶段就关键技术问题提出沟通交流申请,每类会议针对不同情况开展,其中Pre-IND会议属于II类会议。 I类:
II类:
※ 特别注意:对于技术指南明确、药物临床试验有成熟研究经验,申请人能够保障申报资料质量的,或国际同步研发的国际多中心临床试验申请,在监管体系完善的国家和地区已经获准实施临床试验的,申请人可不经沟通交流直接提出临床试验申请。 III类: 除Ⅰ类和Ⅱ类会议之外的其他会议。 02申请沟通交流会会议流程:
(点击查看大图) ※ 注:从申请人申请到召开会议,I类会议需要在30日内,II类会议需60日内,III类会议需75日内开展。 IND受理与审评审批流程
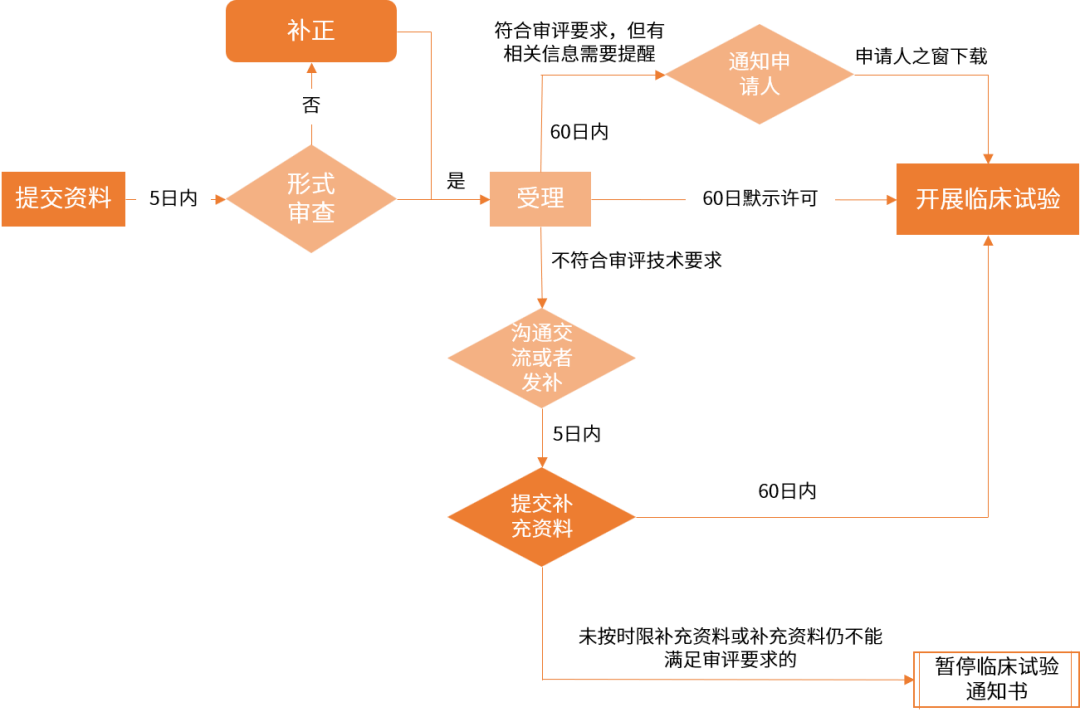
(点击查看大图) IND注册IND注册资料要求 根据国家药品监督管理局《关于调整药物临床试验审评审批程序的公告》(2018年第50号)及原食品药品监管总局发布的关于《新药I期临床试验申请技术指南》的通告(2018年第16号),IND申请需准备的资料包括以下:
介绍性说明和总体研究计划 01介绍性说明: 包括新药的名称、所有的活性成分、药理作用类别、结构式(如果已知)、剂型、制剂处方、给药途径、临床试验目的等。如果有所研究药物用于临床的经验,应提供简短概述,包括在其他国家的研究和上市的经验;若没有,标题下写“无”。 02总体研究计划: 总结申请临床试验方案的设计依据,主要为拟定的适应症、受试者人群、受试者数量、给药方案(剂量、给药间隔、给药持续时间等)、药物安全性评价方法、风险控制计划等,根据已有信息预期的任何安全性(重要的已确定风险、重要的潜在风险、重要的缺失的资料等)的风险论证。 研究者手册 包括封面页、目录、保密声明、概述、新药名称与理化性质、非临床研究结果(药理作用、毒理作用、非临床药代动力学研究)、已有临床研究或使用资料(人体药物代谢动力学、有效性、安全性及上市情况)、其他及参考文献。 临床试验方案 包括研究背景,试验目的,预计参加的受试者数量,入选标准和排除标准描述,给药计划描述,检测指标、对受试者安全性评价至关重要的相关试验详细信息,中止研究的毒性判定原则及试验暂停标准等。 药学研究信息 重点关注对计划研究的受试者安全性相关的药学研究信息,出现以下药学问题应暂缓临床试验:
非临床研究信息
既往临床使用经验说明
作者简介:半夏svt,就职于北京新领先医药,从事药政工作多年,已成功完成多项药品的注册申报工作,拥有丰富的项目经验。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号