前 言 为加强医疗器械临床试验过程的监督管理,指导监管部门开展医疗器械临床试验监督检查工作,根据《医疗器械注册管理办法》和《医疗器械临床试验质量管理规范》要求,NMPA分别于2016年、2018年组织制定了《医疗器械临床试验现场检查要点》《医疗器械临床试验检查要点及判定原则》。 目前,我国医疗器械临床试验质量距离"核查要点"还有一定的差距,申办者、合同研究组织、研究者、临床试验机构都应该进一步学习相关法规,提高临床试验质量,确保上市后的产品安全。本期拟通过解读医疗器械临床试验现场核查要点,以规范医疗器械临床试验过程。 开展医疗器械临床试验的目的是什么? 在医疗器械领域开展临床试验,可达到以下目的: 1、支持医疗器械新产品上市和老产品注册证的更新,为注册提供足够的数据支持; 2、收集市场研究所需的信息和数据(上市前/上市后),支持公司战略; 3、开展医疗器械卫生经济学研究,为物价/医保/招标工作提供证据支持; 4、开展医疗器械临床培训,给希望做研究的医生提供思路和方法,提供临床交流的平台,促进中国医疗器械临床试验的规范及发展; 5、支持研究者发起的研究以及相关科研结果的发表,推动中国医生参与全球交流; 6、在各种临床试验活动中,建立起与医生及医院的长期合作关系,传递公司的学术性,建立起良好的公司形象。 哪些医疗器械的上市申请需要做临床试验? 根据2014年10月1日生效的《医疗器械注册管理办法》(国家食品药品监督管理总局令 第4号),第一类医疗器械实行备案管理,第二类、第三类医疗器械实行注册管理。办理第一类医疗器械备案,不需进行临床试验,申请第二类、第三类医疗器械注册,应当进行临床试验。 对于已列入免于进行临床试验的第二类、第三类医疗器械目录的产品,根据《医疗器械临床评价技术指导原则》提交相应临床评价资料的,可免于进行临床试验。对于未列入免于进行临床试验的第二类、第三类医疗器械目录的产品,如按照《医疗器械临床评价技术指导原则》规定的“通过同品种医疗器械临床试验或临床使用获得的数据进行分析评价”途径能够证明产品的安全性和有效性,亦可不进行临床试验,反之则需要临床试验的数据来支持产品的有效性和安全性。第三类医疗器械由于风险较高,除非在NMPA公布的临床试验豁免清单内,一般需要在申请注册前开展临床试验。
1、在试验前确认临床试验机构已具有适当的条件,包括人员配备与培训符合要求,实验室设备齐全、工作情况良好,预期有足够数量的受试者,参与研究人员熟悉试验要求。 2、 在试验前、中、后期监察临床试验机构和研究者是否遵循有关法规、本规范和临床试验方案。 3、确认每位受试者在参与临床试验前签署知情同意书,了解受试者的入选情况以及试验的进展状况;对研究者未能做到的随访、未进行的试验、未做的检查,以及是否对错误、遗漏做出纠正等,应当清楚、如实记录;对修订的知情同意书,确认未结束临床试验流程并受影响的受试者重新签署。 4、确认所有病例报告表填写正确,并与原始资料一致;所有错误或者遗漏均已改正或者注明,经研究者签名并注明日期;每一试验的病种、病例总数和病例的性别、年龄、治疗效果等均应当确认并记录。 5、确认受试者退出临床试验或者不依从知情同意书规定要求的情况记录在案,并与研究者讨论此种情况。 6、确认所有不良事件、并发症和其他医疗器械缺陷均记录在案,SAE和可能导至SAE的医疗器械缺陷在规定时间内作出报告并记录在案。 7、监察试验用医疗器械样品的供给、使用、维护以及运输、接受、储存、分发、处理与回收。 8、监督临床试验过程中相关设备的定期维护和校准。 9、确保研究者收到的所有临床试验相关文件为最新版本。 10、每次监察后应当书面报告申办者,报告应当包括监察员姓名、监察日期、监察时间、监察地点、监察内容、研究者姓名、项目完成情况、存在的问题、结论以及对错误、遗漏做出的纠正等。
医疗器械临床试验现场核查的重点有哪些方面? 作为医疗器械临床试验的监督管理手段,NMPA已自2016年开始以抽查的方式,对全国范围内开展的医疗器械临床试验进行核查。临床试验现场核查的重点主要包括如下几方面: 1、承担医疗器械临床试验机构的资质情况:是否是GCP或相关临床试验指导原则规定的医疗机构。 2、临床试验伦理审查情况:临床试验是否经伦理委员会审查同意并签署知情同意书。 3、临床试验方案制定与执行情况:例如,临床试验方案是否申请人与临床试验机构协商制订;临床试验方案是否经伦理委员会审查同意;临床试验过程是否遵循临床试验方案开展;各临床试验机构执行的方案是否统一;临床试验方案规定的试验内容是否由试验机构完整开展等。 4、申办者履职情况:例如,是否签署临床试验合同/协议,并明确相关方在临床试验中的责任;是否对试验人员进行培训;临床试验前是否进行预试验;申请人是否派人员对临床试验进行监察等。 5、临床试验记录情况:例如,临床试验过程是否完整、可追溯;临床试验的CER表是否填写完整并与原始资料一致等。 6、试验用医疗器械的管理情况:例如,试验用医疗器械是否有具有资质检测机构出具的合格报告;试验用医疗器械是否与临床试验报告中的产品一致;试验用医疗器械的管理记录(包括分发、运送、接受、回收与销毁)是否完整,数量是否相符等。 7、临床试验申报资料情况:例如,注册申请提交的临床试验方案与试验机构保存的试验方案版本及内容是否一致;注册申请提交临床试验报告中的数据与原始记录中的相应数据是否一致;注册申请提交临床试验报告中载明的样本量与各临床试验机构实际承担的样本量是否一致;临床试验报告上的临床试验人员签名是否属实,其他签章是否符合要求等。 2020年新增和修订的免于进行临床试验医疗器械目录(征求意见稿) 2020年7月9日,国家药监局器审中心网站发布《2020年新增和修订的免于进行临床试验医疗器械目录(征求意见稿)》(厅字〔2017〕42号),旨在进一步规范医疗器械临床评价工作,扩大免于进行临床试验的医疗器械目录范围,并面向社会公开征求意见,截止至2020年8月31日可在器审中心网站下载意见反馈表,对目录提出意见和建议。
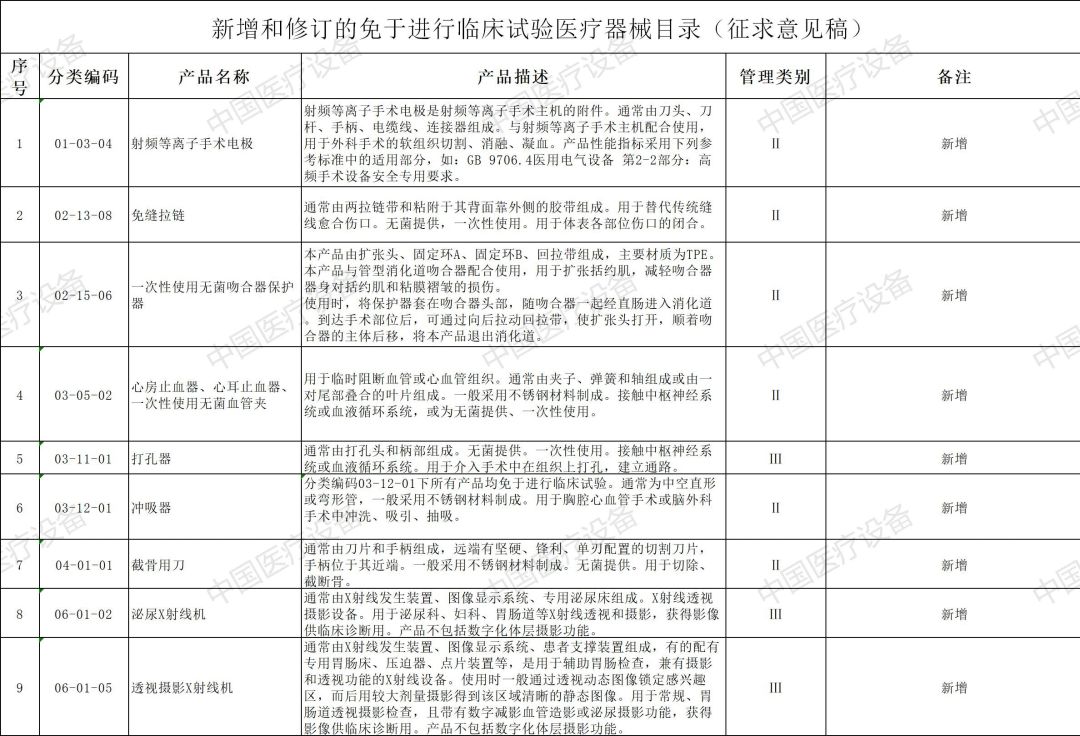
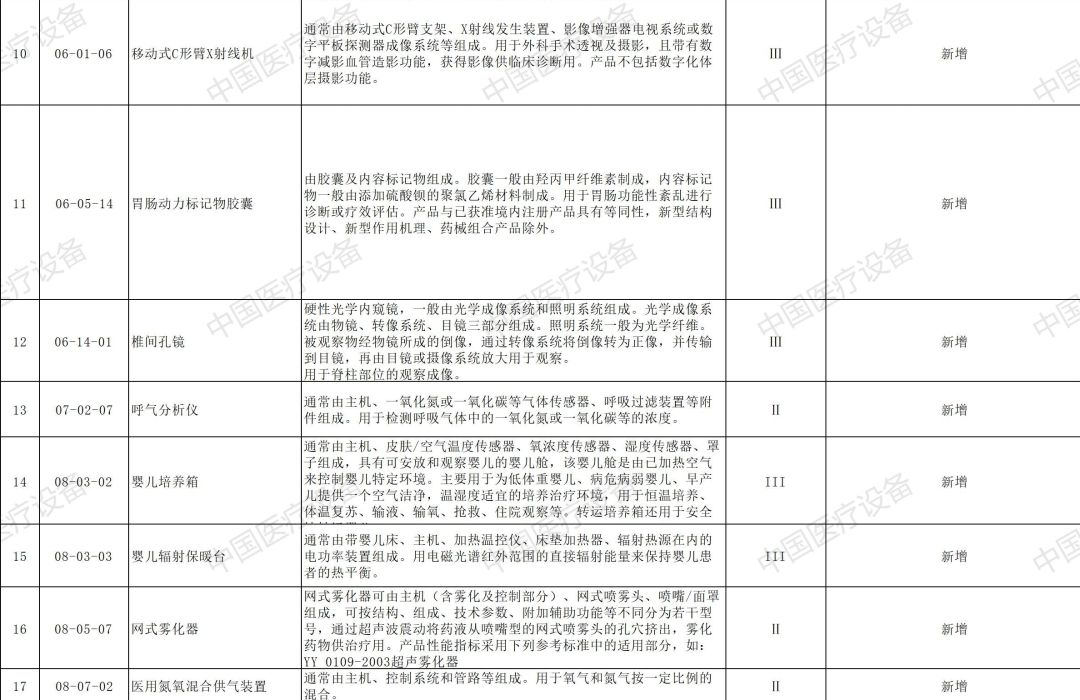
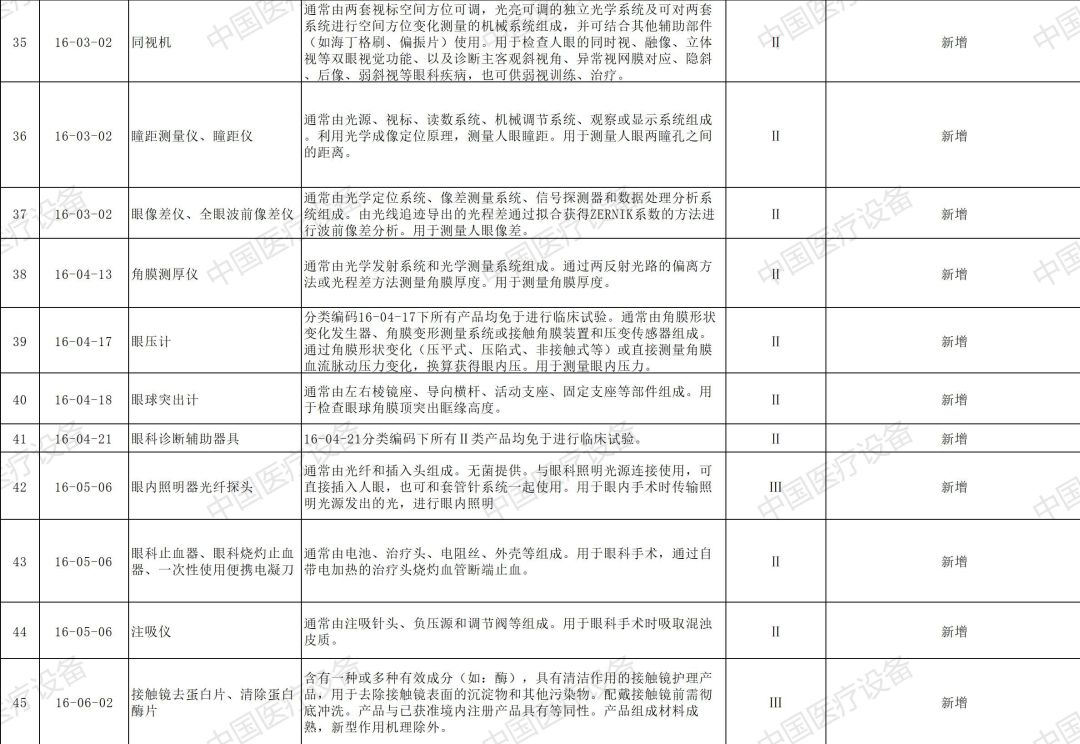
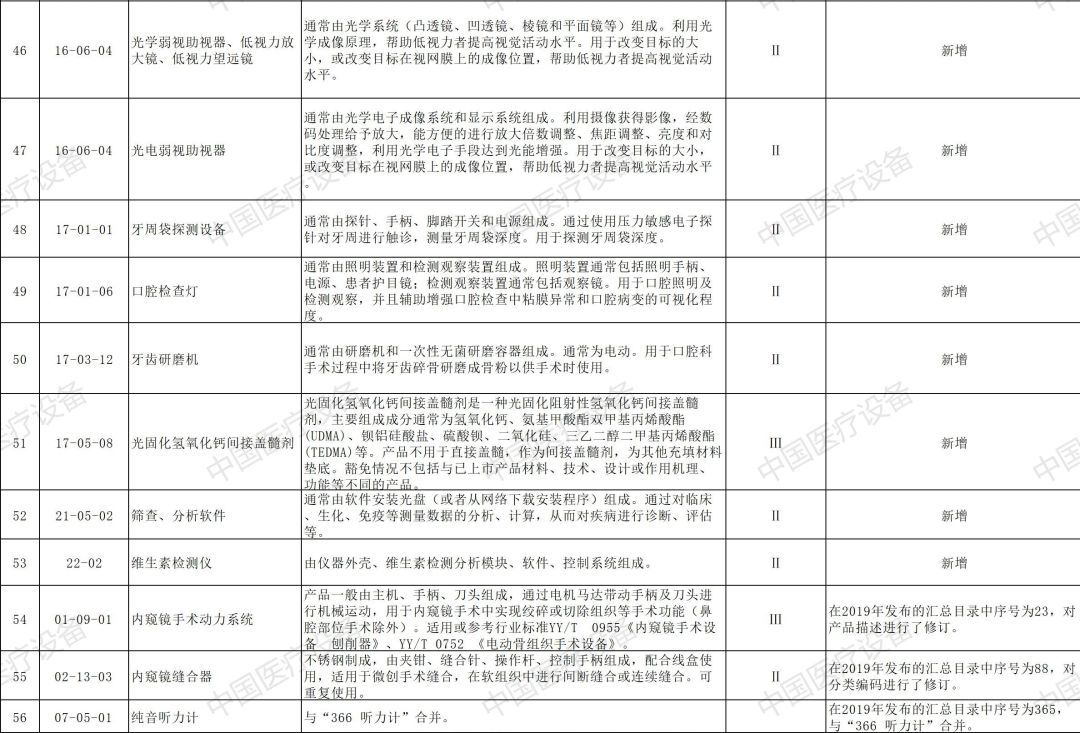
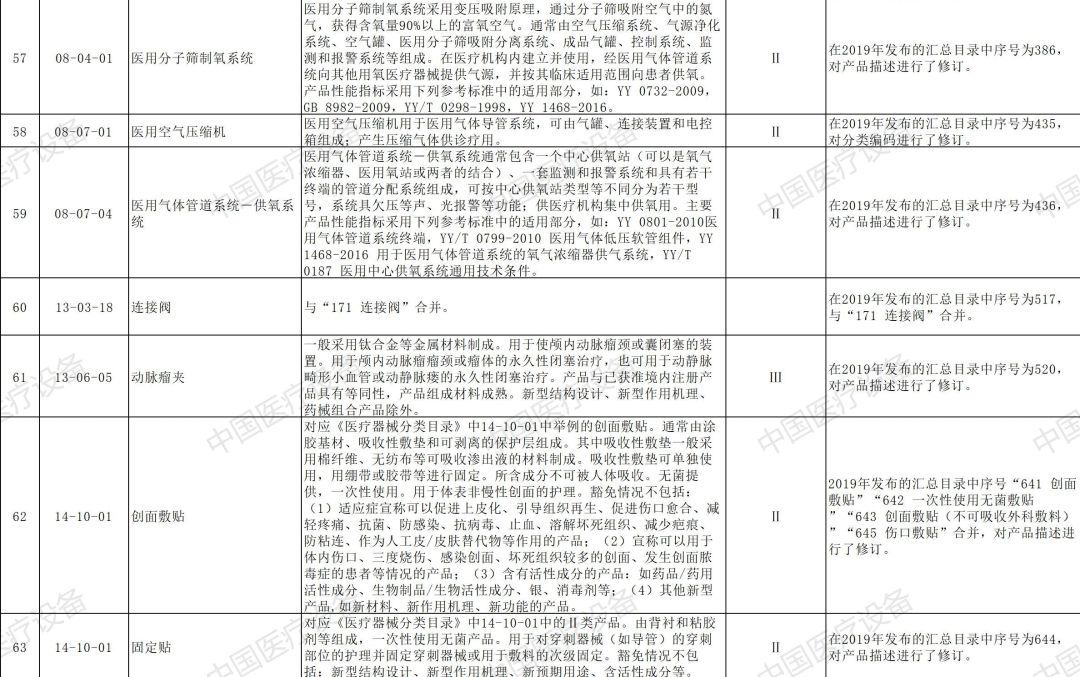
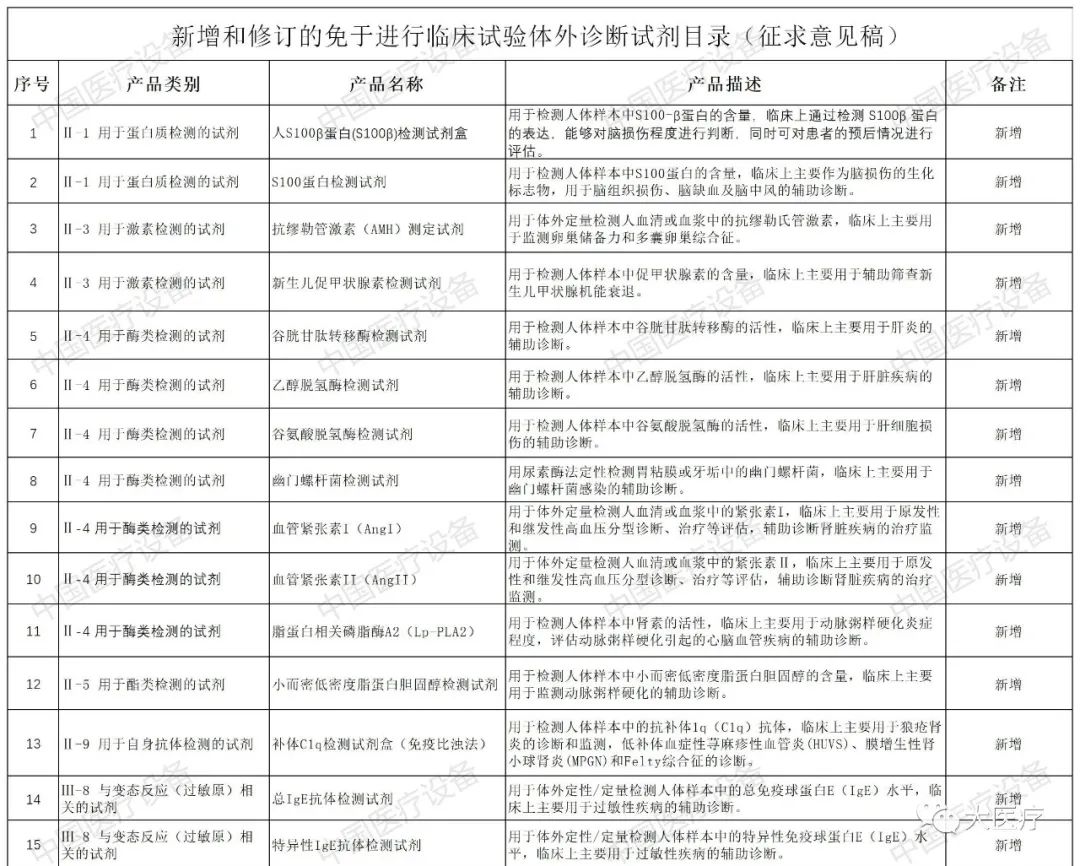
以下是2020年新增和修订的免于进行临床试验医疗器械目录(征求意见稿)与2020年新增和修订的免于进行临床试验体外诊断试剂目录(征求意见稿)详情:
- End - |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号