人类体外辅助生殖技术用液(以下简称ART用液)是指与人类配子和/或胚胎发生直接或间接接触,以准备、培养、转移或存储人类配子和/或胚胎的液体类医疗器械产品,如取卵液、卵子与胚胎处理液、精子洗涤液、精子密度梯度分离液、体外受精液、卵裂胚培养液、配子/胚胎冷冻液、配子/胚胎解冻液、培养用油等。目前,此类产品在我国被作为第三类医疗器械进行管理。 近年来,我国辅助生殖技术迅速发展,每年需消耗大量的辅助生殖培养液等耗材。同时,国家药品监督管理局医疗器械技术审评中心(以下简称器审中心)收到的ART用液相关产品的注册申报数量不断增加。 为指导产品研发及注册申报,国家药监局于2018年4月发布了《人类体外辅助生殖技术用液注册技术审查指导原则》。
目前,国内已上市ART用液均为进口产品,部分国产产品已进入临床评价阶段。近两年来,器审中心多次接到境内外企业有关此类产品注册申报方面的咨询,问题集中反映在以下方面:由于此类产品的特殊性,根据《医疗器械临床评价技术指导原则》按同品种比对路径进行临床评价存在困难、临床试验研究病例入组有难度。国内企业因缺少产品实际应用的数据,临床评价困难问题更为突出。 基于以上原因,器审中心深入调研了美国、欧盟、澳大利亚、日本等国家和地区监管机构对ART用液产品监管、注册申报、临床评价等方面的要求,以期为解决此类产品在我国注册申报时存在的临床评价方面的问题提供经验,支持产品的国产化。
一、其他国家和地区的监管及临床评价要求 1、美国 ART用液在美国被作为Ⅱ类医疗器械进行管理,通过提交上市前通告[510(k)]的途径上市,即通过实质等同途径,便可获得上市申请并免于提交临床评价资料。 FDA于2004年发布了《用于体外受精和相关辅助生殖手术的器械:510(k)提交指南》,根据该指南文件及FDA官网公布的相关信息,在对此类申报器械与合法上市的同品种器械进行比对时,FDA所要求的比对内容主要包括:产品的预期用途、技术特征(配方、pH值、渗透压、细菌内毒素、除菌方式、鼠胚试验结果),以及申报器械的生物学评价资料。 在配方方面,FDA未要求提交所有成分的浓度对比信息。并且,FDA允许将申报产品与多个已合法上市的产品进行对比,以证明申报产品与同品种产品的差异并非代表新的技术,两者实质等同,具有临床安全性和有效性。 在对是否需进行临床试验的规定上,FDA指南文件明确指出:“对用于辅助生殖过程的器械,临床试验并非一项常规要求。然而,对于一些性能结构设计或功能与现有辅助生殖过程常规器械不同的产品,可能需要进行额外的临床试验,以充分验证、评估产品性能,如,具有特殊设计的胚胎移植导管,生产商宣称含特殊成分、能够提高妊娠率的培养液等。” 2、欧盟 欧盟于2012年发布了《体外受精和辅助生殖技术产品的合格评价指南》,关于ART用液的管理类别,该文件指出:“在辅助生殖过程中与生殖细胞密切接触的用于清洗、分离、精子制动、冷冻保护的溶液为Ⅱb类,用于支持胚胎生长/储存的培养液、含有人血清白蛋白(HSA)和/或抗生素等药物成分的产品、含有动物组织或衍生物成分的产品均为Ⅲ类。由于ART用液类产品普遍含有HSA及抗生素成分,在欧盟相关规定中,除培养用油为Ⅱa类,某些不含有HSA及抗生素成分的精子密度梯度分离液为Ⅱb类,其他产品均按Ⅲ类产品进行管理。 ART用液在欧盟国家上市需提供符合规定的临床评价资料,即通过实质等同评价路径来提供临床评价报告或临床试验资料。欧盟的实质等同路径与我国的同品种比对路径的基本原则一致,包括实质等同性判定、提供实质等同器械的临床安全有效性证据。但是,欧盟与我国的做法有一定区别。我国在进行同品种比对路径临床评价时,要求提供申报产品的临床数据,以证明差异性对产品安全性和有效性的影响,还要求提供所比对的同类产品的临床数据,以证明所申报产品具有安全性和有效性。欧盟要求提供的临床安全性评价证据基本上是同类产品的文献数据的总结。 关于临床研究的必要性,欧盟指南文件规定:对缺乏技术或经验的高风险医疗器械,可能需要提供临床研究数据;而是否进行额外的临床研究取决于现有临床数据是否能证明产品符合基本要求。部分企业的调研资料显示,生产商必要时会对申报ART用液产品进行临床试验,以支持其在欧盟的上市申请。 3、澳大利亚 ART用液在澳大利亚的管理类别与欧盟一致。澳大利亚治疗产品管理局(TGA)于2008年发布了对此类产品上市申请安全性及有效性进行评估的申报资料要求,包括产品技术指标要求(pH值、渗透压、细菌内毒素、杂质含量,以及鼠胚试验、遗传毒性等生物学项目)。在临床评价资料方面,TGA要求提供实质等同途径的临床评价报告。 4、日本 ART用液在日本不属于医疗器械,而是按照实验室试剂管理。
二、审评思考与建议 器审中心在充分调研并借鉴其他国家和地区对ART用液类产品监管及上市申报、临床评价规定的基础上,结合我国法规及相关产业发展的需要,针对此类产品在临床评价及技术审评中存在的问题提出几点建议。 (一)扩充《免于进行临床试验医疗器械目录》
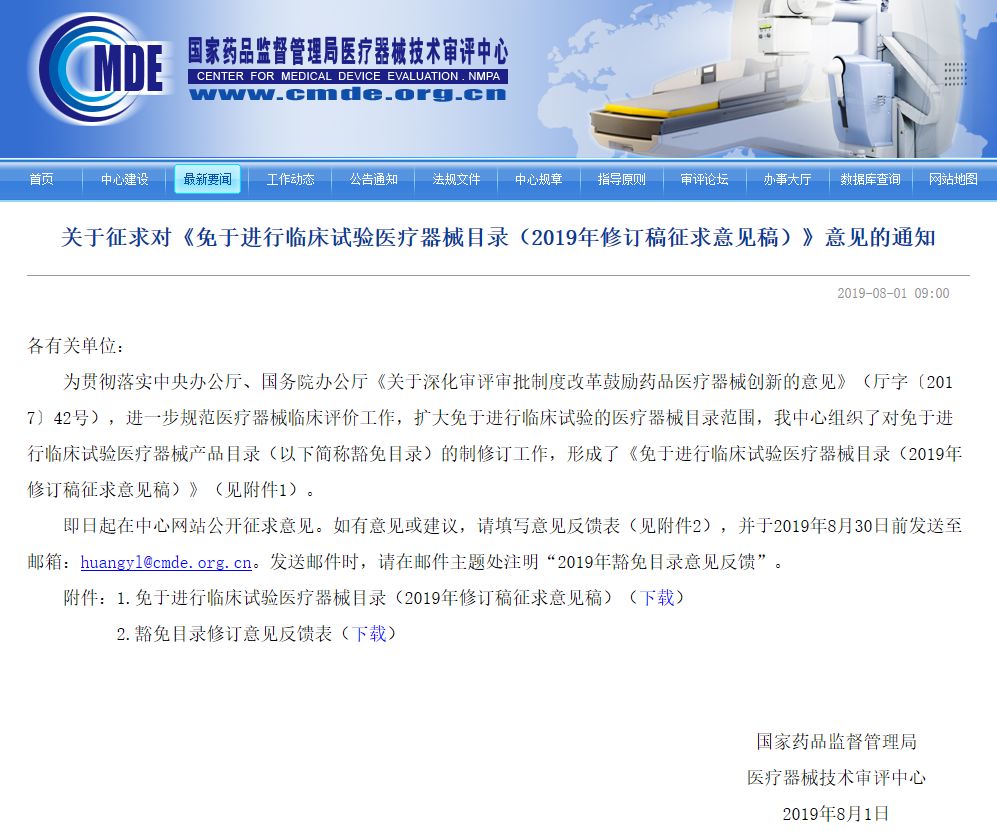
现阶段,培养用油已被纳入《免于进行临床试验医疗器械目录》。建议根据产品的技术成熟度及临床风险可控度,选取部分产品纳入下一批目录。在2018年8月CMDE发布《免于进行临床试验医疗器械目录》(2019年修订稿征求意见稿)中,精子密度梯度分离液、卵泡冲洗液等部分技术相对成熟、短时接触及风险程度较低的产品可被优先考虑。
▲2018年8月CMDE发布《免于进行临床试验医疗器械目录》(2019年修订稿征求意见稿)拟新增人类辅助生殖技术用液项目(来自:CMDE) (二)简化同品种路径审评方法 在对此类产品的注册审评中,企业反映较多的问题是,由于申报产品与已上市同品种产品在组成成分上存在差异,难以获得同品种产品各组分浓度数据,导至通过同品种比对路径提交临床评价资料的可操作性较差。对此,器审中心借鉴FDA等机构的审评要求,建议允许将所申报产品与多个同品种产品的组成成分进行比对,以证明所申报产品不含有新的组成成分,对无机盐等常见基础组分,不再强制要求提供浓度对比信息。 基础组分浓度差异对安全性、有效性的影响可通过性能指标参数(如pH值、渗透压、杂质限量、使用性能指标等)的对比来体现。对与预期用途相关的特殊功能性组分,需提供浓度对比信息,并评价差异性对产品安全性和有效性的影响。 (三)需进行临床试验的情形 结合产品风险程度,器审中心建议,对以下几种情形,需要通过开展临床试验来进一步验证产品的安全性和有效性。 1.产品含有国内已上市同类产品中未含有的新的组成成分。 2.产品的预期用途宣称与常规的国内已上市产品不同,如可改善精子活力、提高妊娠率等。 3.产品的主要成分浓度发生重大调整,而无充分的临床安全有效性支持资料。 (四)提出上市后随访要求 在国产产品首次获批上市时,有必要对其提出上市后随访要求,随访指标主要包括活产率、出生健康情况等。
以上建议基本涵盖ART用液类产品目前在我国进行注册申报时面临的临床评价中的所有难点。这些问题的解决可加快国产产品上市进程,促进我国自主研发的ART用液类产品更快地走向市场。 生命的诞生是一个神圣而精妙的过程,ART用液类产品直接作用于人类生殖细胞和胚胎,对此类产品的质量控制应比对普通医疗器械的质量控制标准更严格。在技术审评中,审评主体除严格按照法规及相关指导原则来审评各项非临床及临床评价资料外,还应严格审查产品质量体系相关文件,规范并强化风险管理,以促进体外辅助生殖技术的健康发展。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号