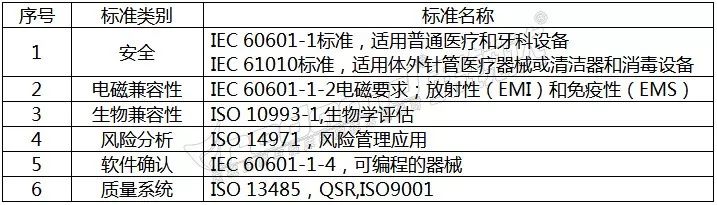
医疗器械标准
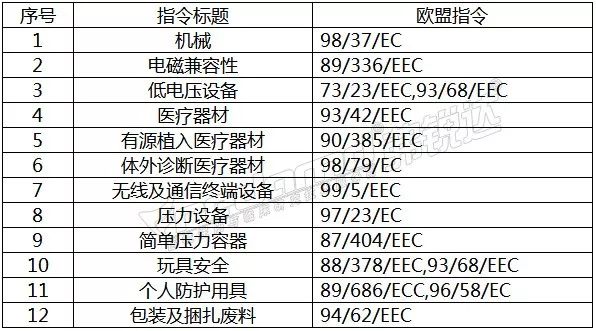
医疗器械指令列表
欧盟
所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的以下相关指令:
2017 年5 月25 日,MDR和IVDR 正式生效, 替代了原医疗器械指令(MDD,93/42/EEC)、有源植入医疗器械指令(AIMD,90/385/EEC)和体外诊断试剂医疗器械指令(IVDD 98/79 EC),并分别于2020 年5 月26 日和2022 年5 月26 日强制实施。 医疗器械指令(MDD),适用于大多数进入欧盟销售的医疗设备。它根据不同的要求共分为6个等级,供认证机构评估。
体外诊断医疗器械指令(IVDD)可按以下分类申请:
北美地区
在美国,食品和药物管理局(FDA)是监督和管理获准向消费者进行销售的食物,药物,化妆品和医疗器械的法定机构。器械及放射线健康中心(CDRH),作为FDA的一个分支,专管医疗器械。其对医疗器械按不同等级进行不同程度的监管(医疗器械分为I级,II级或III级,I级作为低风险范畴,而III级属高风险范畴):
在加拿大方面,加拿大医疗器械认证认可机构(CMDCAS)要求医疗器械厂商提前获得经CMDCAS认可的第三方机构,如UL的质量体系审查,证明其质量系统符合CMDCAS的ISO13485/ISO13488标准。对CMDCAS认证的了解对于完成FDA的质量系统注册(QSR)非常有帮助,因为如上所述的QSR是以ISO 9001和ISO 13485标准为基础的。 制造商有责任获取和验证已被肯定的器械的相关信息,比如目录,使用说明书和510(k)其它要求的资料信息。
通常,有三种情况需要申请510(k):
FDA 510(k)审查 从2002年10月1日起,审查需直接向FDA缴纳用户费。经过FDA的初次审查,申请人将收到FDA出示的产品缺陷报告或声明,这个过程通常需时90天。经过改正和/或其它资料的补充后,FDA随后还将再进行为期90天的复审。 要缩短510(k)审查的周期,并减少工作量,第三方510(k)审查是完成审核的另一选择。如您选择诸如UL这样的第三方评审机构,那么整个审查可在四周内完成。
日本
日本的保健体系和美国的完全不同。日本政府制定了严格的产品认证流程,新进入日本市场的外国医疗设备产品都必须严格遵守。为了进入日本市场,医疗产品厂商必须首先获得两种由日本厚生省(MHLW)颁发的文件——营业执照和上市许可证。外国厂商必须委托一个在日本已取得营业执照的代理商。国外企业和日本国内的代理商同时负责适用于其产品的进口程序和文件、GMP标准和售后监督的认证工作。在日本,产品根据不同风险程度(由低到高)分为3类。UL根据日本国内标准如JIS T1001和JIS T1002为客户提供“类型测试(Type Testing)”服务
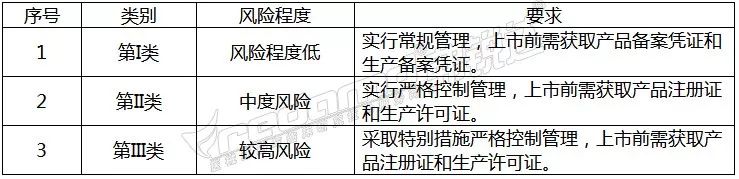
中国
中国的医疗器械由国家市场监督管理总局监督管理,按照《医疗器械监督管理条例》规定,按医疗器械的风险程度不同,分为以下三类:
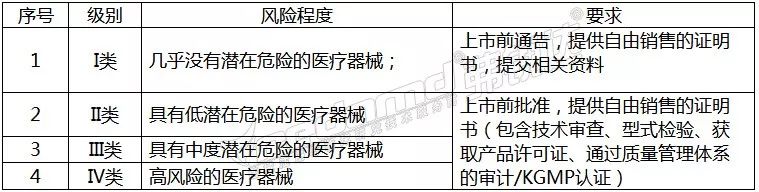
韩国
韩国卫生福利部(MinistryofHealthandWelfare,MHW),简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是主要的卫生保健部门。依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部(MinistryofFoodandDrugSafety,MFDS)负责对医疗器械的监管工作。 韩国医疗器械分类方法与欧盟对医疗器械的分类方法非常相似,分为以下四类:
澳大利亚
澳大利亚的I、Ⅱa、Ⅱb、Ⅲ和AIMD五类医疗器械需由医疗器械管理机构为治疗品管理局(TGA,Therapeutic Goods Administration)的准许才能获得上市准入。生产商向TGA递交申请上市材料后,按照符合性审查程序进行审核。TGA要求医疗器械制造商的质量体系符合ISO13485:2003医疗器械质量体系的要求,认可欧盟的CE认证。由TGA对制造商的质量体系进行审核。 澳大利亚对于上市的医疗器械产品实施警告系统和事故报告机制,通过采用包括不良事件的调查报告、上市产品的实验室检验和监测活动保证其符合法规的规定。要求赞助人和制造商应该将所有的不良事件信息汇报给TGA。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号