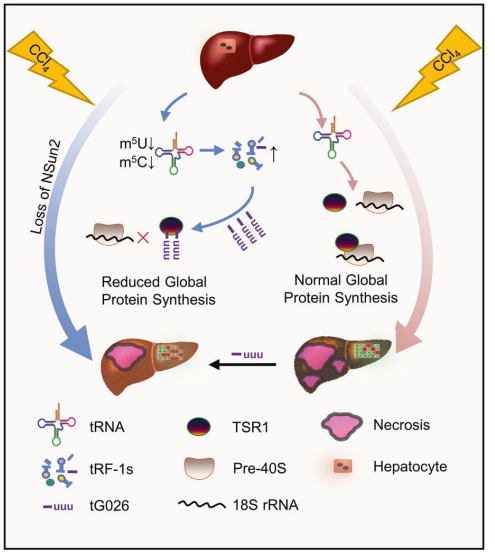
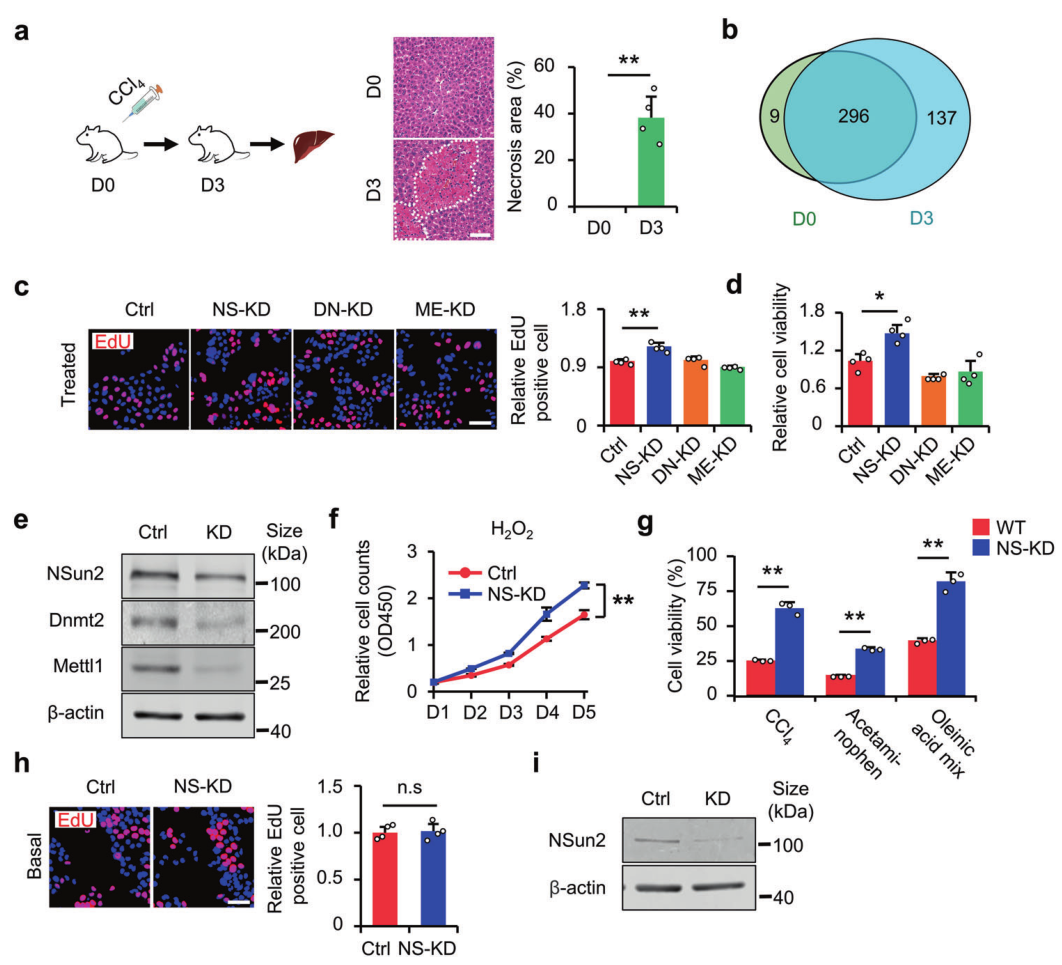
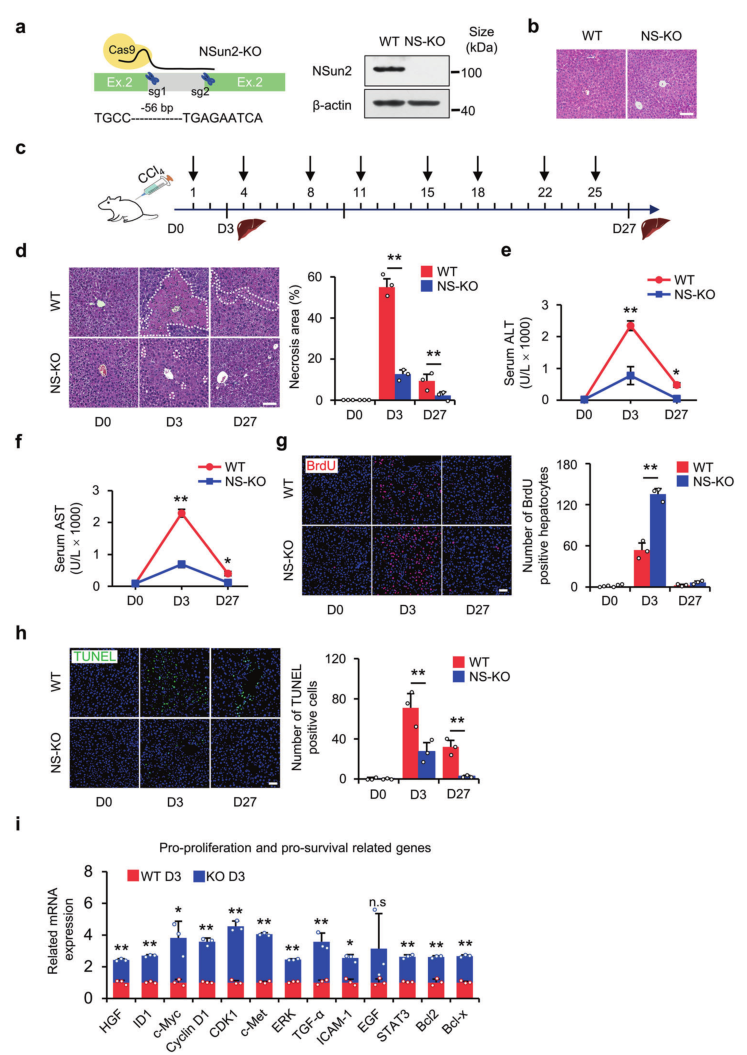
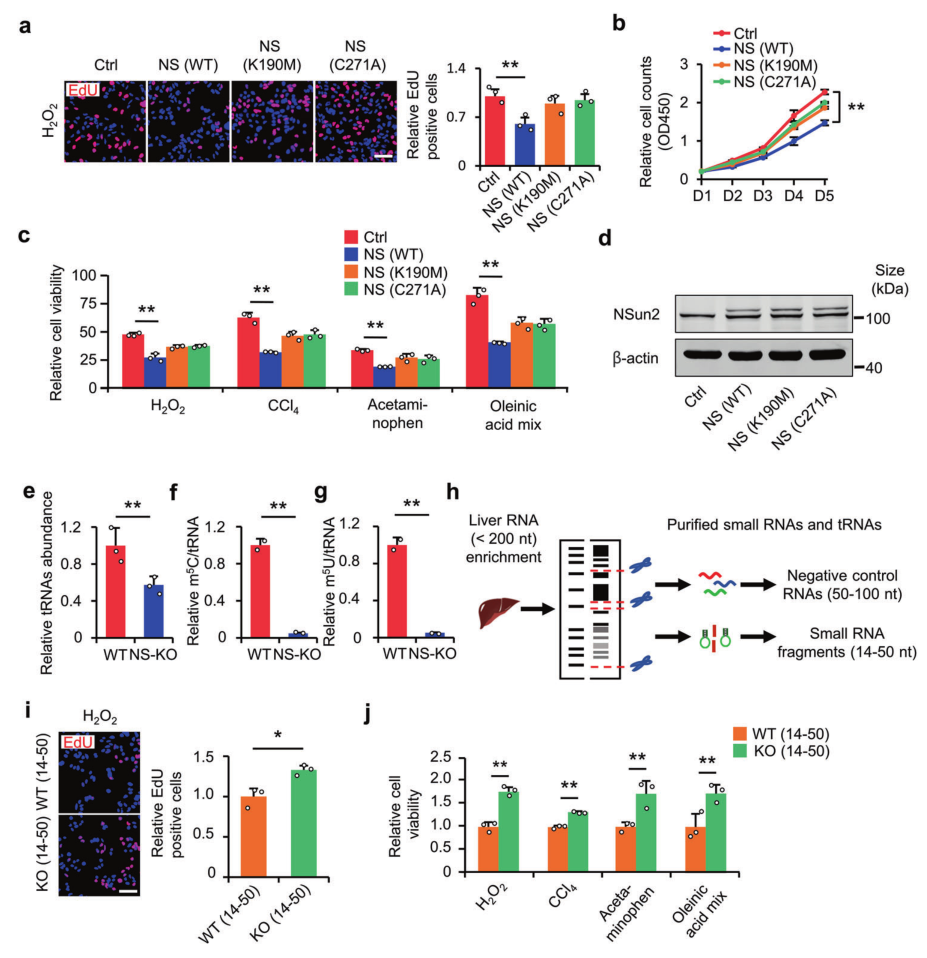
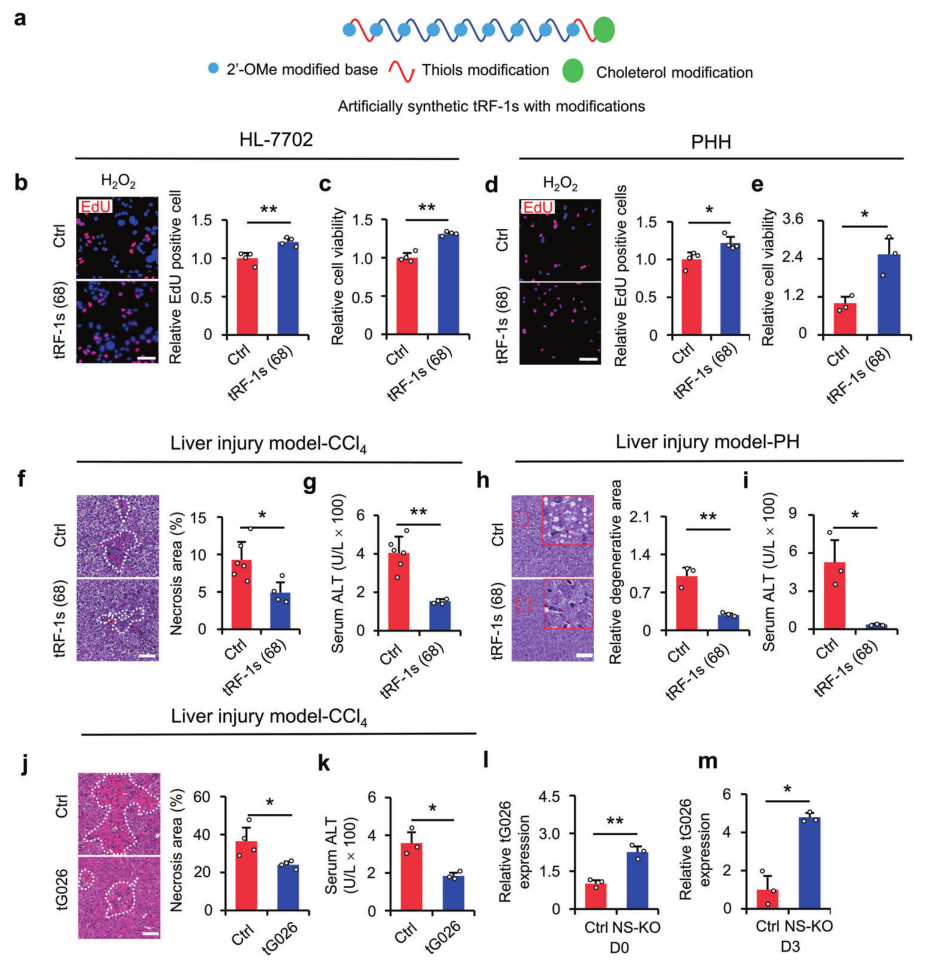
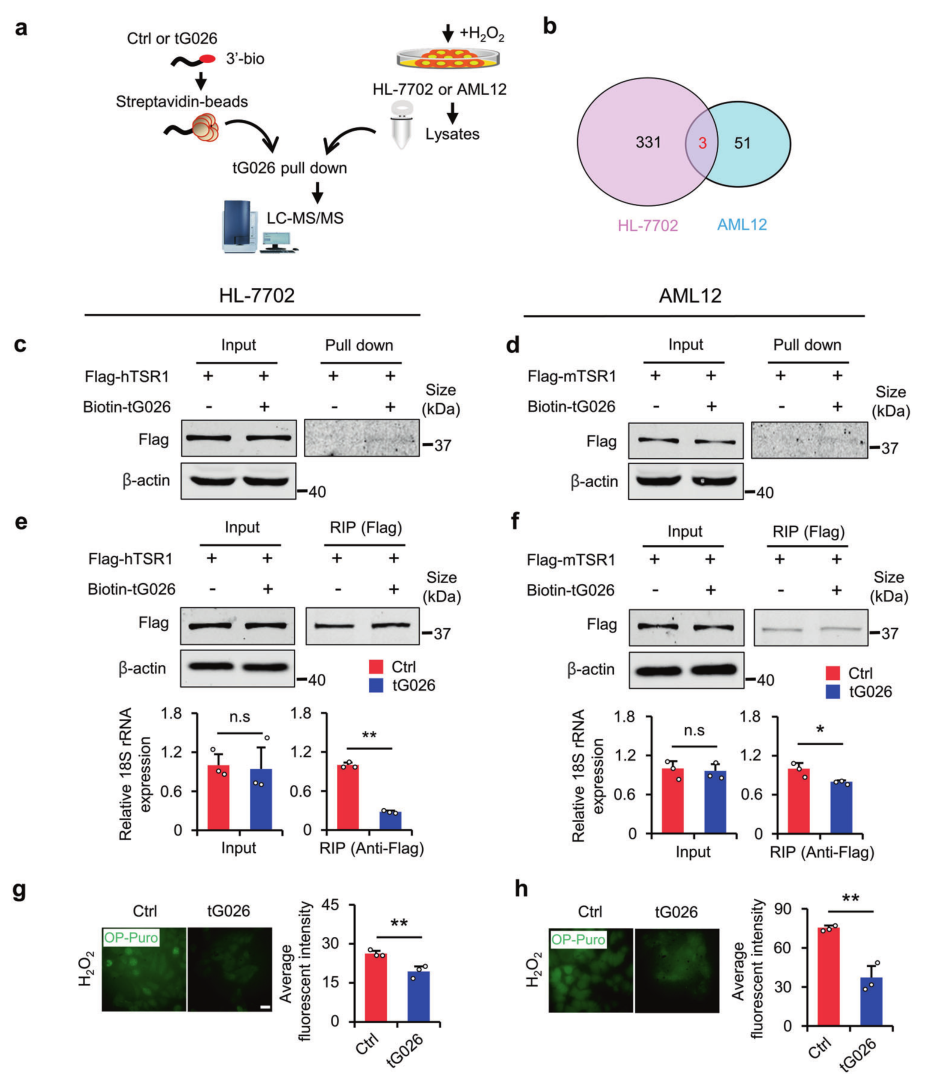
金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
一 ABPP 技术介绍
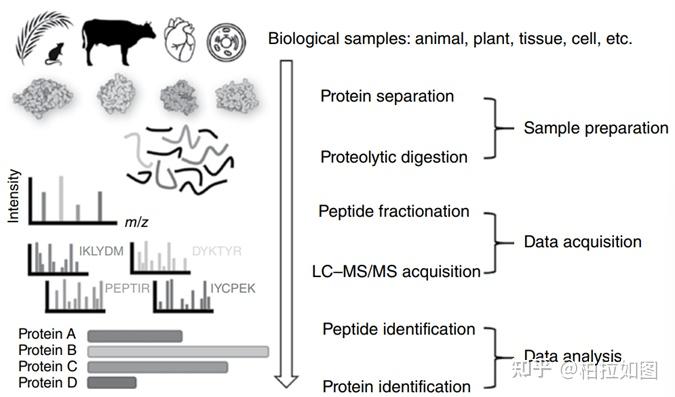
基于活性的蛋白质谱(activity-based protein profiling,ABPP)是一种化学蛋白质组学方法,它结合基于活性的探针(activity-based probe,ABP)和蛋白质组学技术来鉴定活性小分子的蛋白质靶标,以帮助阐明活性小分子发挥作用的机理。
基于 ABPP 技术的靶蛋白垂钓技术可以分为以下三个步骤:
1. 活性分子探针设计合成及活性检测
2. 利用改造后的活性小分子垂钓靶蛋白并进行鉴定
3. 活性小分子与靶蛋白间的相互作用验证。
在早期的研究中,活性分子常被共价固定在生物相容的惰性载体上,但这种共价固定往往会损伤活性分子的生物活性。为了克服上述的缺点,科学家开发了基于活性的探针(ABPs),对活性小分子进行标记,报告标签在被引入活性小分子时不会影响小分子的活性。
ABPs 由三个部分组成:活性反应基团、链接基团和报告标签。活性反应基团是在活性分子与靶点蛋白结合后,通过反应将整个分子以共价键的形式结合到蛋白上,目前常用的活性反应基团为光反应基团(如双吖丙啶);链接基团是在活性分子和报告标签之间创造足够的空间来保持活性分子的活性;报告标签则是用来检测或纯化与活性分子结合的靶蛋白(如生物素)。
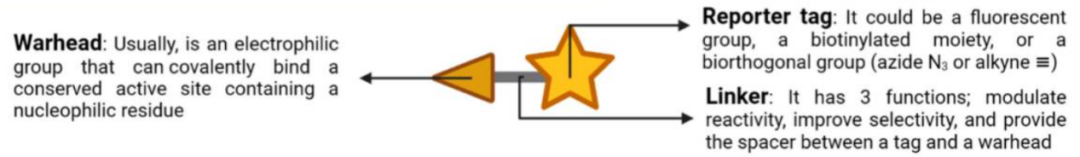
图1 ABPS 结构示意图
在 ABPP 技术早期,报告标签通常直接与活性小分子相连,导致整个探针的分子量过大,不利于 ABPs 通过细胞膜进入细胞。随着点击化学的发现,生物正交反应的发展给 ABPP 技术带来了长足的发展。给活性分子引入更小的炔烃修饰,等活性分子进入细胞后,再通过点击化学反应对蛋白配体复合物中的配体进行生物素化修饰,从而可以进行进一步的靶蛋白富集鉴定。
二 ABPP 主要实验步骤
1. 探针制备:活性分子探针的设计合成,以及探针活性鉴定
2. 将探针与总蛋白孵育(或与细胞孵育后提取总蛋白)
3. 上述孵育完成后,先后进行 UV 照射(使活性分子与靶蛋白共价相连)、点击化学反应(给活性分子引入报告基团,如生物素)
4. 通过报告基团对靶蛋白进行富集鉴定
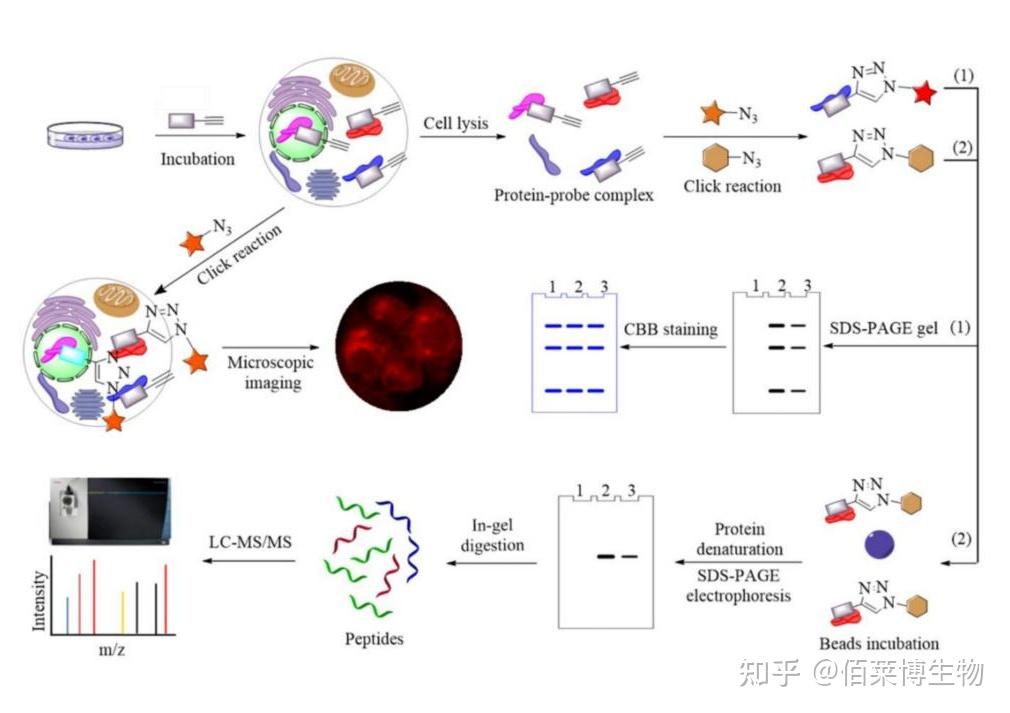
图2 ABPP 实验基本流程图
三 案例介绍
案例一:基于 ABPP 技术的绿原酸(Chlorogenic acid)靶蛋白鉴定[1]
研究者将光报告分子-双吖丙啶连接到 CGA 上。实验时,将 PAL/CGA 探针与线粒体蛋白混合进行孵育,同时平行组加入未标记的 CGA(作为对照),孵育完成后,使用 UV 进行照射,使光亲和分子与靶蛋白之间形成牢固的共价键,随后通过点击化学反应给探针连上生物素标记,最后通过链霉亲和素富集靶蛋白并通过质谱鉴定。经质谱鉴定,线粒体乙酰辅酶A乙酰转移酶1(ACAT1)可能是 CGA 的直接作用靶点。该研究借用 ABPP 技术筛选到了绿原酸(CGA)潜在蛋白靶点 ACAT1,并通过 SPR(表面等离子共振)、ITC(等温滴定量热)、TSA(热迁移)以及 DARTS(drug affinity responsive target stability)等实验进一步证明了 CGA 与 ACAT1 蛋白的相互作用。
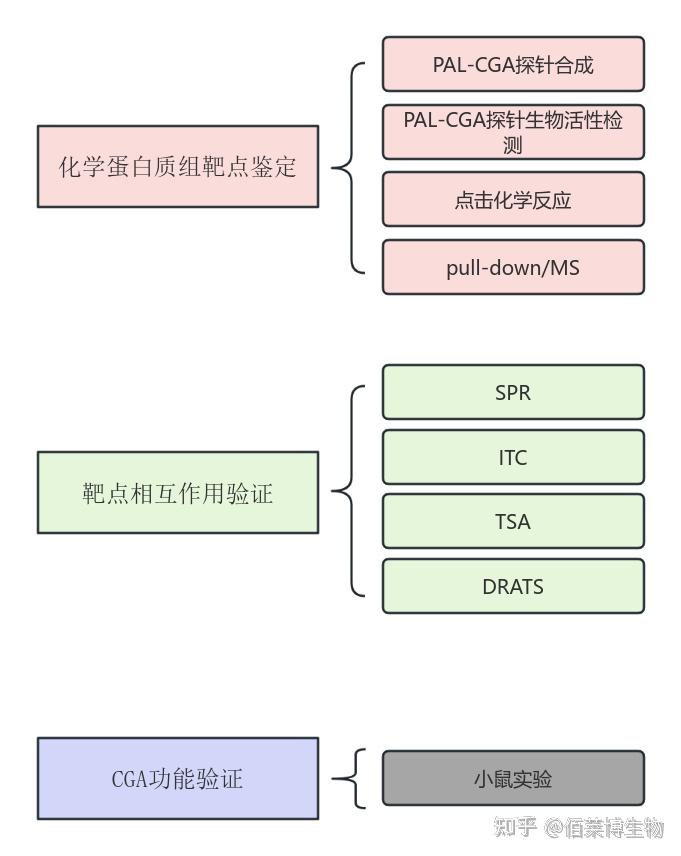
图3 本研究思维导图
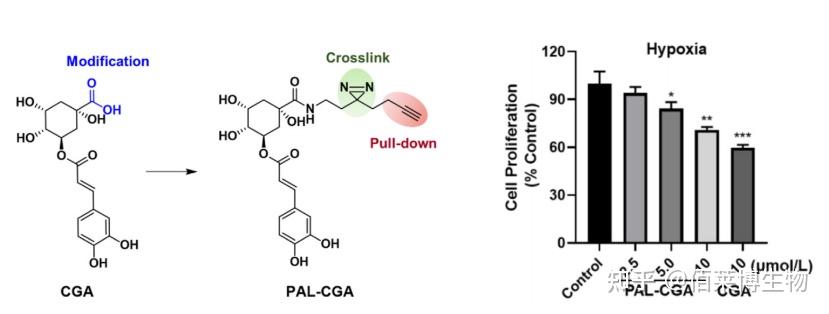
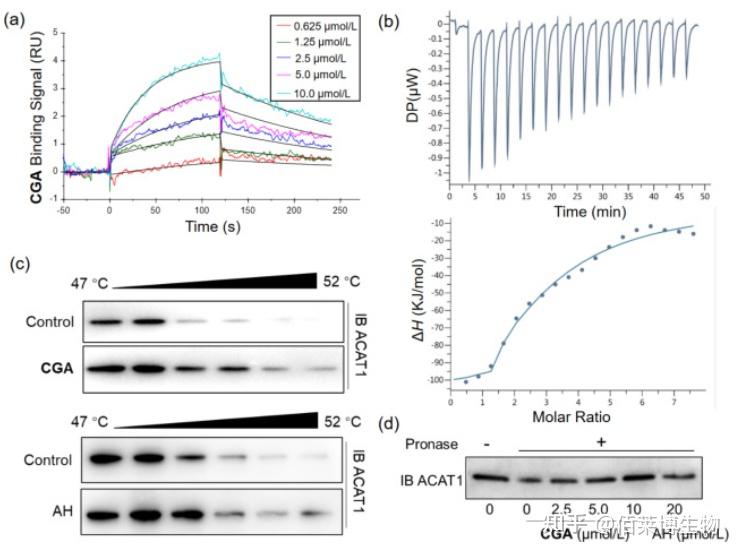
图4 本研究活性探针结构及 SPR(表面等离子共振)、ITC(等温滴定量热)、TSA(热迁移实验)和 DARTS(drug affinity responsive target stability)证明 CGA 和 ACAT1 蛋白的相互作用
Photoaffinity purification
CGA was used as a control to compare with PAL/CGA, a photoaffinity labeling probe. CGA was incubated with mitochondrial proteins at 50 mmol/L, while PAL/CGA was added at 10 mmol/L, both at 4 ℃ for 2 h. Subsequently, the samples were exposed to UV light (365 nm) at 4℃ for 30 min, followed by the addition of azide biotin at 20 mmol/L. A catalyst, composed of CuSO4 (20 mmol/L) and TECP (25 mmol/L), was used to reduce Cu(II) to Cu(I), and THPTA (60 mmol/L) was added to stabilize Cu(I). After 18 h of rotation at room temperature, proteins were precipitated with cold acetone, washed thrice, and dissolved in 1% SDS in PBS. PAL/CGA-bound proteins, isolated using streptavidin beads (Invitrogen), were eluted, subjected to SDS-PAGE, and visualized with Coomassie blue staining. Bands differing in abundance between the CGA-preincubated and control samples were analyzed by LC‒MS/MS using a Q Exactive Mass Spectrometer (Beijing Protein Innovation, China). Protein identification was performed using Mascot Daemon (version 2.3.0).
案例二:基于 ABPP 技术解析由 FBP 及其靶蛋白 ALDH2 介导的存在于线粒体途径的糖代谢信号感知机制[2]
研究者将光报告分子-双吖丙啶连接到 FBP 上。实验时,将 FBP 探针与细胞进行孵育,孵育完成后,使用 UV 进行照射,使光亲和分子与靶蛋白之间形成牢固的共价键,随后对细胞进行裂解,用点击化学反应给探针连上生物素标记,最后通过链霉亲和素富集靶蛋白并通过质谱鉴定。经质谱鉴定,线粒体代谢酶-乙醛脱氢酶2(ALDH2)可能是 FBP 的直接作用靶点。通过分子对接(molecular docking)、MST(微量热泳动)、小分子免疫荧光、小分子pull-down 等实验进一步验证了 FBP 和 ALDH2 之间的相互作用。并通过活细胞实验进一步阐明了 FBP 介导的糖代谢信号感知机制。
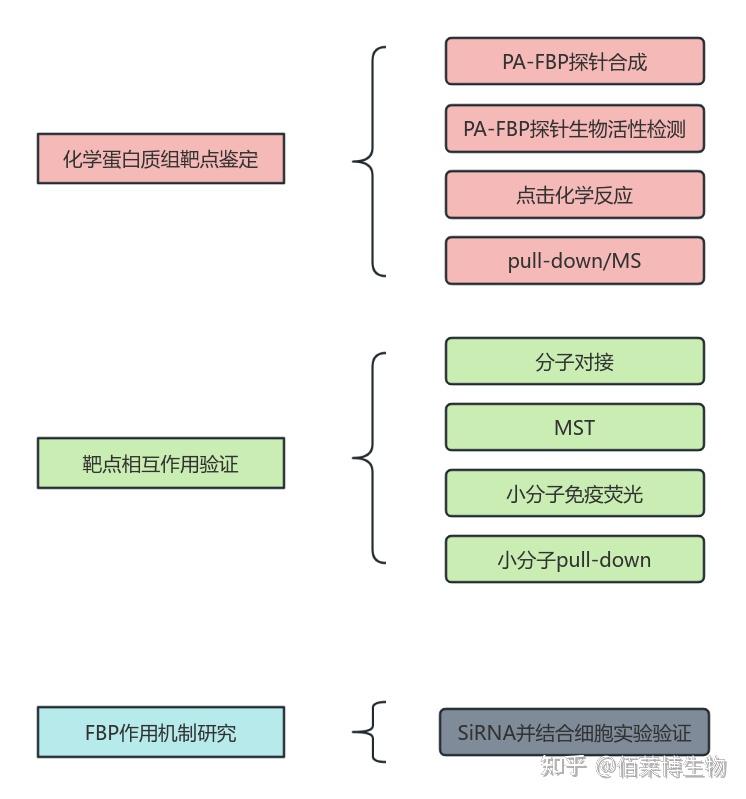
图5 本研究思维导图
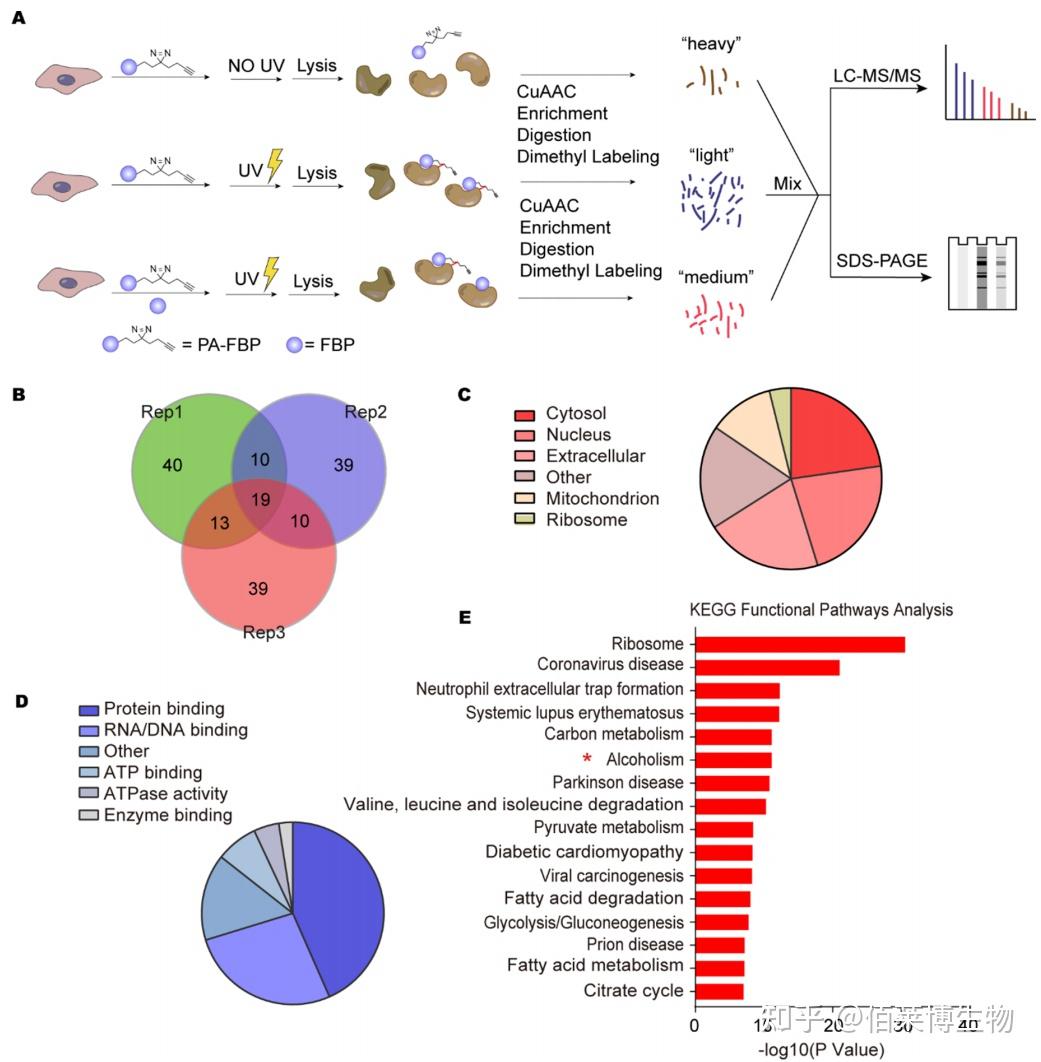
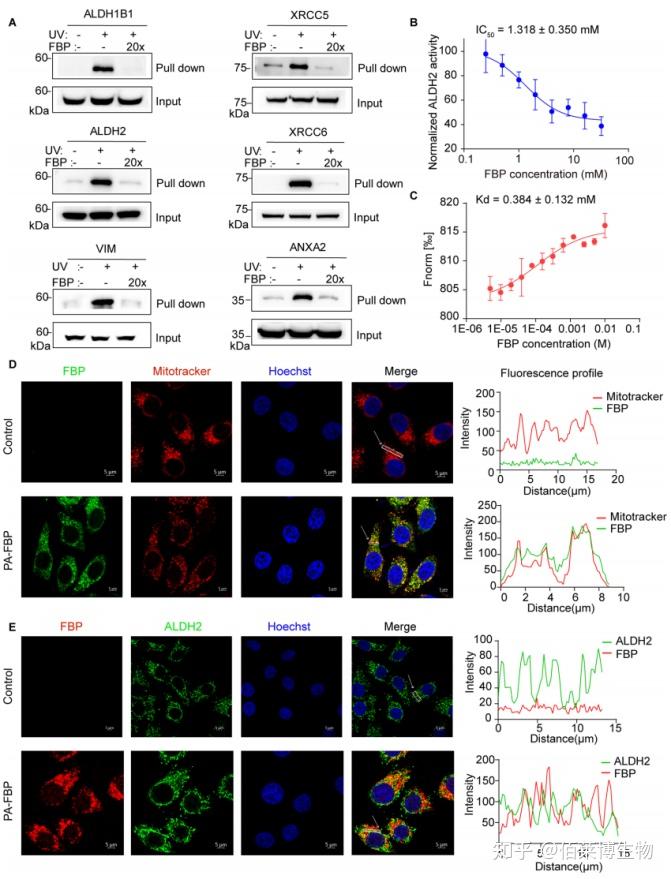
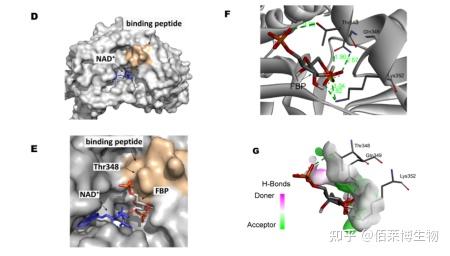
图6 本研究 ABPP 基本实验流程图及分子对接(molecular docking)、MST(微量热泳动)、小分子免疫荧光、小分子pull-down 验证 FBP 和 ALDH2 存在相互作用
Quantitative chemoproteomic profiling of FBP-interacting proteins in living cells
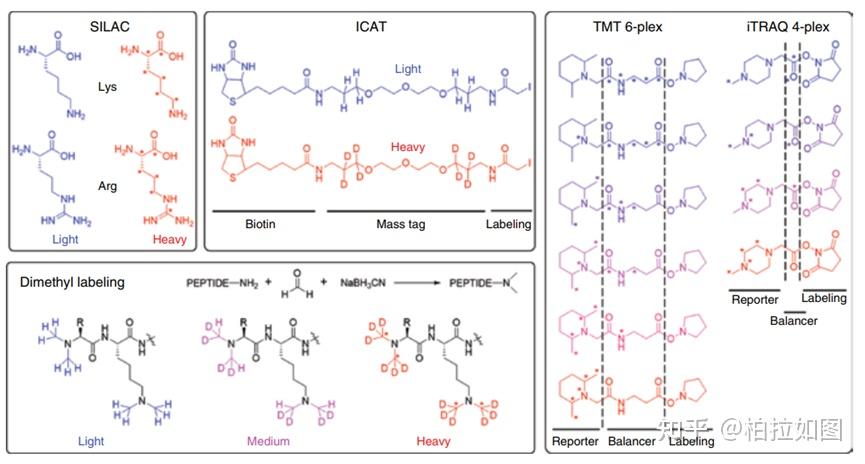
For the profiling of FBP-interacting proteins in living cells, HepG2 cells were seeded into a 15-cm dish (20 × 106 cells per plate) overnight with DMEM (Gibco, C11995500CP) containing 10% fetal bovine serum (Excell), 1% Penicillin/Streptomycin (Gibco, 15140122). The growth medium was aspirated, and the cells were washed with PBS gently for three times. The cells were treated with 1 mM PA-FBP for 90 min with or without UV light irradiation (365 nm, 5 min), and 1mM PA-FBP with excess FBP (TargetMol, USA, T8074) for 90 min with UV light irradiation (365 nm, 5 min). The cells were washed with PBS. Cells were then dislodged by scraping in PBS, pelleted by centrifugation at 500 g for 5 min at 4 ℃. The cell pellets were stored at -80 ℃. The cells were lysed by sonication in ice with ice-cold 0.1% TritonX-100/PBS buffer and cell lysates were collected by centrifugation (20,000 g, 30 min) at 4 ℃ to remove the debris. The protein concentration was determined by using the PierceTM BCA Protein Assay kit (Thermo Fisher Scientific). 1 mL cell lysates (2 mg/mL) were reacted with 1 mM CuSO4, 100 µM TBTA ligand, 100 µM Biotin-PEG4- amino-t-Bu-DADPS-C6-azide (1260247-50-4), and 1 mM TCEP for 1 h at room temperature. The resulting click-labeled lysates were precipitated by 4000 µL methanol, 1000 µL chloroform and 3000 µL Milli-Q water and then washed twice with 1 mL cold methanol. The proteins were resuspended in 1 mL PBS containing 1.2% SDS. 100 µL streptavidin beads (Thermo Fisher Scientific) were washed for three times and resuspended in 5 mL PBS, which was added to the protein solution. The resulting solution was incubated for 4 h at 29 ℃, followed by washing with 5 mL PBS for three times, and 5 mL distilled water for three times. The resulting beads were resuspended in 500 µL PBS containing 6 M urea and 10 mM DTT and incubated at 37 oC for 30 min, followed by addition of 20 mM IAA for 30 min at 37 oC in the dark. The beads were then collected by centrifugation and resuspended in 200 µL PBS containing 2 M urea, 1 mM CaCl2 and 0.5 µg Lys-C and 2.5 µg trypsin (Enzyme & Spectrum). Trypsin digestion was performed at 37 oC with rotation 16 h and the beads were washed with 200 µL distilled water for three times. For dimethyl labeling, per 100 µL peptides were reacted with 4 µL of 4% “light” formaldehyde (CH2O) (Sigma) and 4 µL of 0.6 M Sodium cyanotrihydridoborate (Sigma), “Medium” formaldehyde (CD2O) (Sigma) and 4 µL of 0.6 M Sodium cyanotrihydridoborate, “heavy” formaldehyde (13CD2O) (Sigma) and 4 µL of 0.6 M Sodium cyanoborodeuteride (Sigma), respectively. The resulting solution was incubated at room temperature for 2 h. The reaction was quenched by adding 16 µL 1% ammonia and 8 µL 5% formic acid. Samples from the “light”, “medium” and “heavy” groups were 1:1:1 mixed and the mixture was collected and desalted with C18 tips (Thermo Fisher Scientific). Eventually, samples were submitted and analyzed by LC-MS/MS using Orbitrap Fusion™ Lumos™ (Thermo scientific, USA). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD048495 (https://www.ebi.ac.uk/pride/).
四 参考文献
[1] Wang QH, Du TT, Zhang ZH, Zhang QY, Zhang J, Li WB, Jiang J-D, Chen XG, Hu H-Y. Target fishing and mechanistic insights of the natural anticancer drug candidate chlorogenic acid, Acta Pharm Sin B 2024; https://doi.org/10.1016/j.apsb.2024.07.005.
[2] Li, Tian & Wang, Anhui & Zhang, Yanling & Chen, Wei & Guo, Yanshen & Yuan, Xia & Liu, Yuan & Geng, Yiqun. (2024). Chemoproteomic Profiling of Signaling Metabolite Fructose-1,6-Bisphosphate Interacting Proteins in Living Cells. Journal of the American Chemical Society. 146. 10.1021/jacs.4c01335. |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-5-11 16:22
发表于 2025-5-11 16:22