金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
近忙了点啥

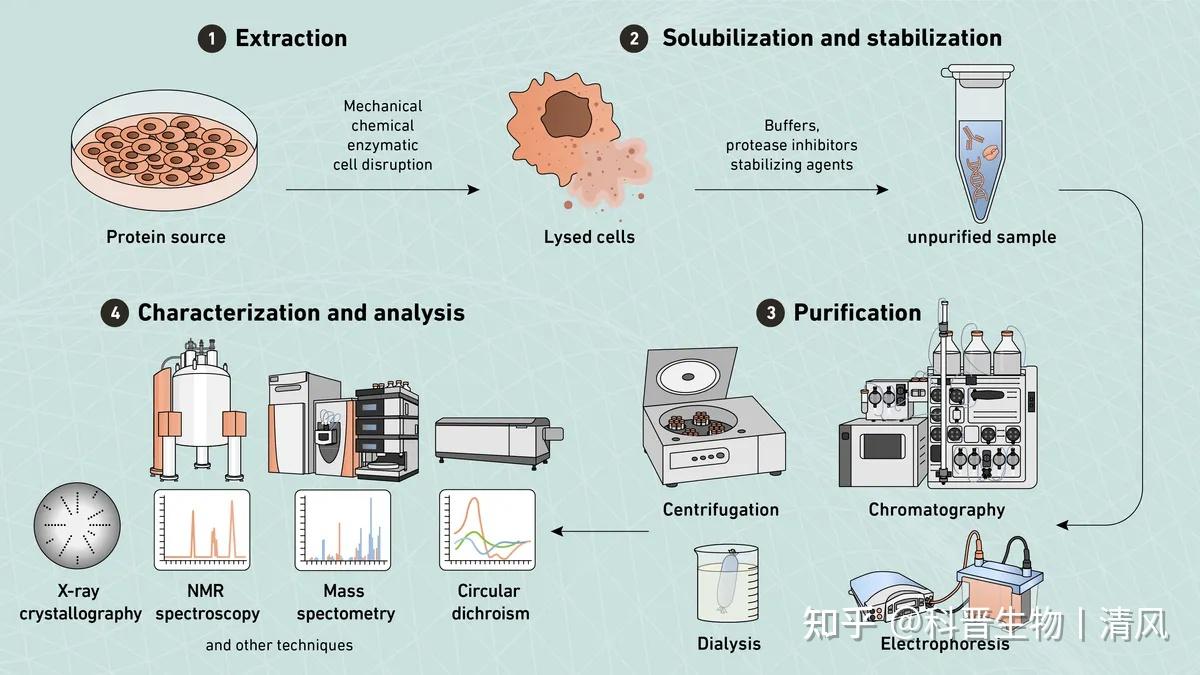
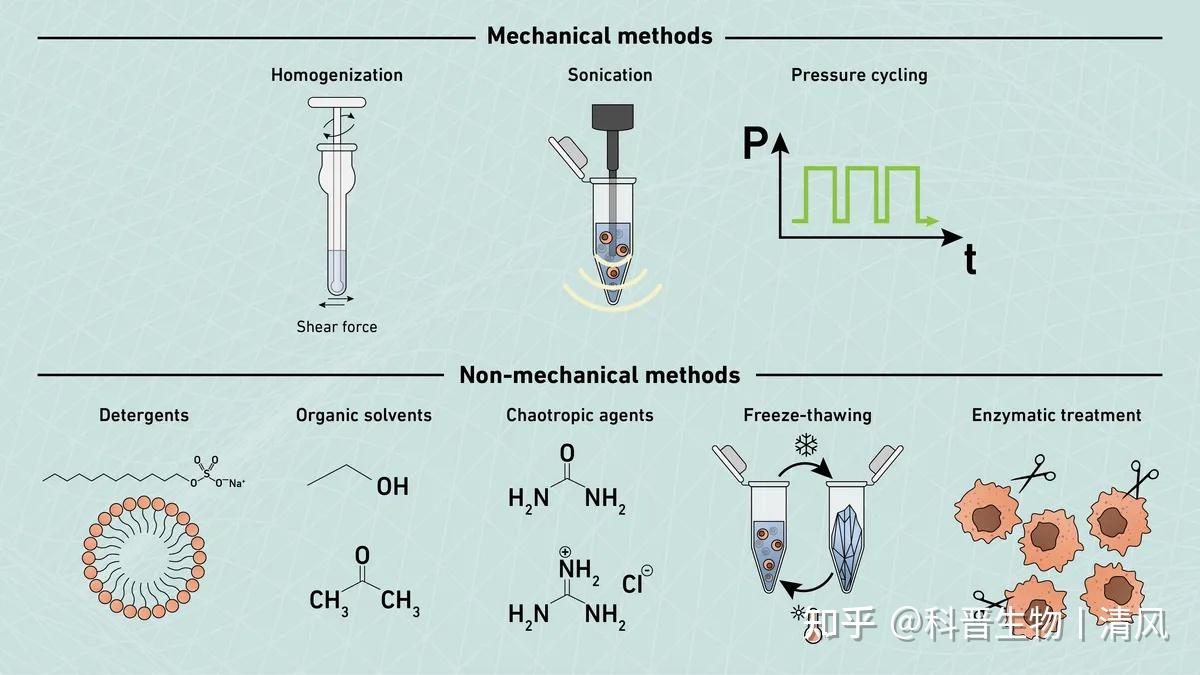
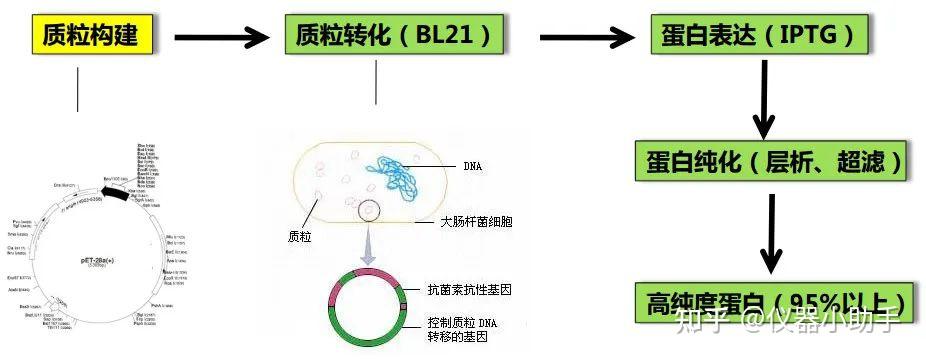
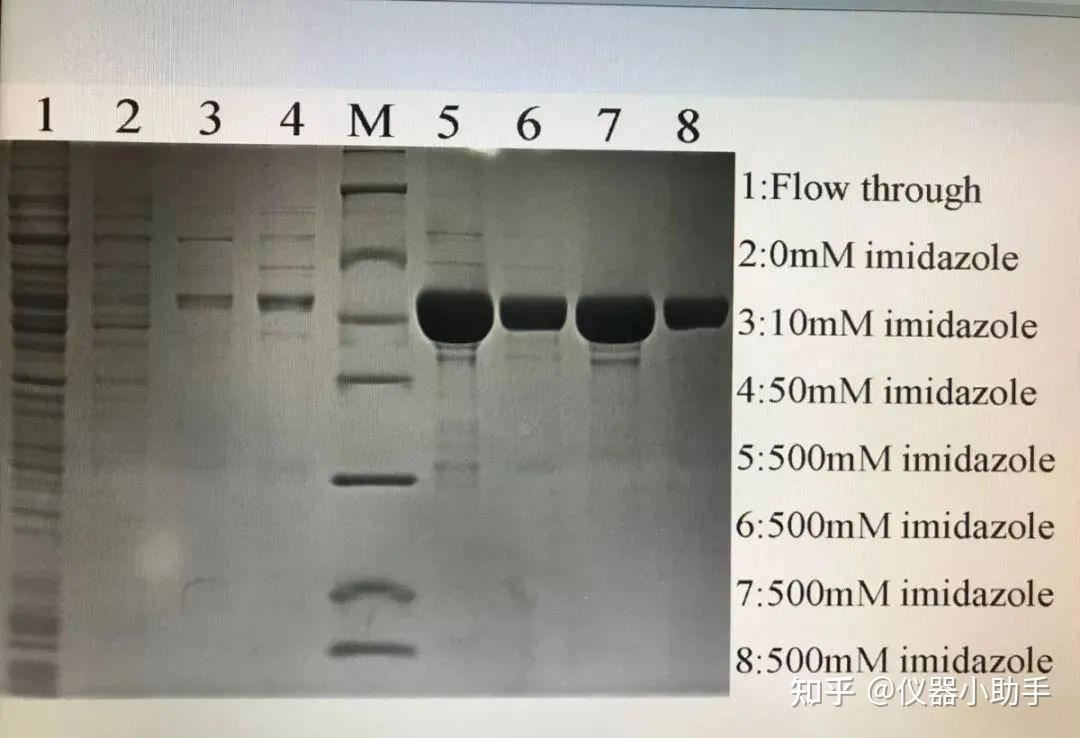
鸽了一周哦,自由散漫惯了,要不也不会喜欢干销售这一行,当然鸽的这段时间也不是闲着,是研究车去了,从年前研究到3月底,终于买了人生的第一辆爱车,一台小小卡罗拉哈哈哈。趁着买车的热乎劲,可以出一期最近看车的心得,感兴趣的还是老规矩投票哈。最后我们根据读者投票,我们先将离子交换层析啦。
基本原理
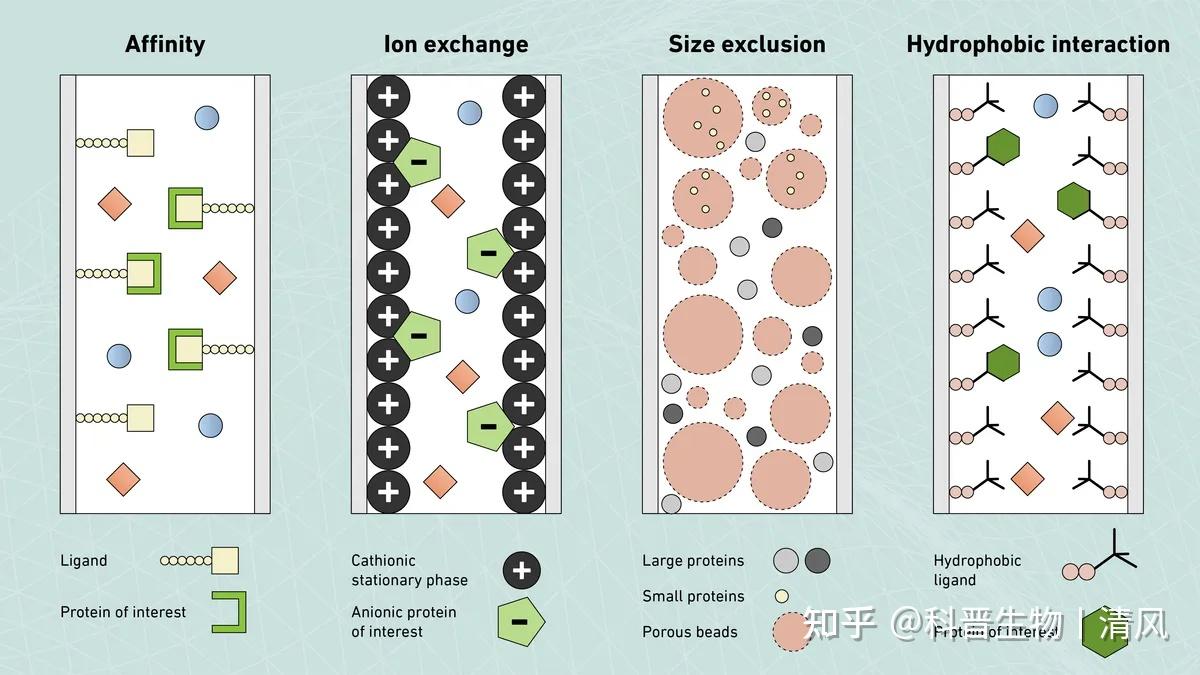
离子交换层析是目前最常用的蛋白纯化技术之一。它利用蛋白和离子之间的电荷相互作用来分离混合物中的不同蛋白。在离子交换层析中,通常使用带有负电荷的离子交换树脂作为固定相,而待纯化的蛋白质溶液作为流动相。当蛋白质溶液通过树脂层时,蛋白分子会根据其表面电荷与树脂上的负电荷发生相互作用,从而被吸附在树脂上。
纯化本质关键
离子交换层析的关键在于,不同蛋白质由于其等电点的差异,通过改变流动液的pH值或加入电解质,可以控制蛋白质与树脂的结合力度,实现不同蛋白质的分离。例如,在低pH值条件下,蛋白质表面电荷较大,与树脂结合力强,此时通过增加pH值可以降低结合力,实现第一组蛋白质的洗脱;而第二组蛋白质由于等电点较高,在此pH下结合力仍较强,需进一步增加pH值才能实现第二组蛋白质的洗脱。通过多次循环改变pH值,就可以实现混合蛋白质的逐步分离。
如何判断蛋白质在某一PH溶液中的电荷量
只需比较蛋白质的等电点PI和溶液的PH值,如果等电点PI大于溶液的PH,那么蛋白质带正电,如果PI小于PH,则蛋白质带负电。交换柱分为阴离子柱和阳离子柱,阴离子柱是为了吸附带负电的蛋白质,而阳离子注是为了吸附带正电的蛋白质。
结合到交换柱上的蛋白质如何取下来
采用盐溶液中的离子和交换柱的蛋白质进行交换,使其脱离,带电荷量少的先脱离,并通过不断加大盐溶液的浓度,洗脱其他带电荷量更高的蛋白质。
恢复与再生
离子交换树脂作为工作材料,在长期使用过程中,其交换位点会逐渐饱和,从而影响其交换效率。因此,对饱和树脂进行恢复或再生成为必要的步骤。
通常有两种方法对离子交换树脂进行处理:一是通过热水或弱酸洗涤,清除吸附在树脂表面或孔隙中的离子,实现树脂的恢复;二是通过强酸或强碱处理,破坏吸附键,剥离深层结合的离子,完全再生树脂。相比恢复,再生能更彻底清除离子,使树脂恢复最初的交换能力。
在操作上,恢复较为简单,主要通过循环冲洗实现。再生需要控制酸碱浓度和时间,防止损害树脂结构,科学合理地进行恢复或再生,对保证离子交换层析系统高效运行至关重要
总结
离子交换层析具有操作简单、成本低廉的优点,在小规模蛋白质初步纯化和中间纯化环节中应用广泛。但是,其分离效率和分辨能力相对较低,难以实现高纯度的蛋白质分离。因此,在后续精细纯化环节,通常还需要结合其他纯化技术,如亲和层析、exclusion层析等进行多级纯化,才能得到高纯度的单一蛋白产物。 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-5-30 16:15
发表于 2025-5-30 16:15


 提升卡
提升卡