登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
分享 | CADD之分子动力学的简介(上篇)引言
分子生物学家一直在试图了解蛋白质或其他生物分子是怎样运作的。原子级别的结构非常有用,通常可以让我们深入了解生物分子的功能。然而,生物分子中的原子是不断运动的,分子功能和分子间的相互作用都取决于所涉及分子的动力学。实验上, 尽管X射线衍射、核磁共振等高分辨结构生物学方法能够给出蛋白质/核酸天然态结构的高分辨原子位置信息, 但通常不能直接提供分子功能运动的动态信息。相反地, 以光谱学技术为代表的各种生物物理方法以及单分子实验技术能够给出分子功能运动的动态信息, 却很难同时提供功能运动过程中高分辨的原子位置等结构信息。为了解决这一难题,研究人员便研发了一种新的方法,用原子级别的计算机模拟相关的生物分子,即分子动力学(MD)模拟。
MD模拟是基于控制原子间相互作用的一般物理模型,预测蛋白质或其他分子系统中的每个原子是如何随时间变动。这些模拟可以捕捉各种重要的生物分子过程,包括构象变化、配体结合和蛋白质折叠,揭示所有原子在飞秒时间分辨率的位置。重要的是,这样的模拟还可以预测生物分子在原子水平上对突变、磷酸化、质子化或配体的添加/去除等任意干扰的反应。特别是近年来, 由于计算机技术的高速发展, 以分子模拟为主的理论方法已成为研究蛋白质等生物大分子功能运动的主要手段之一。
一、MD是什么?
MD的基础
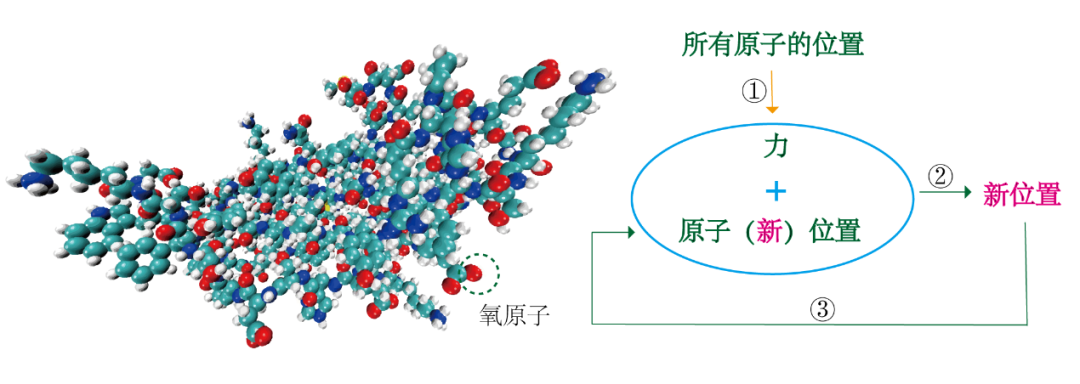
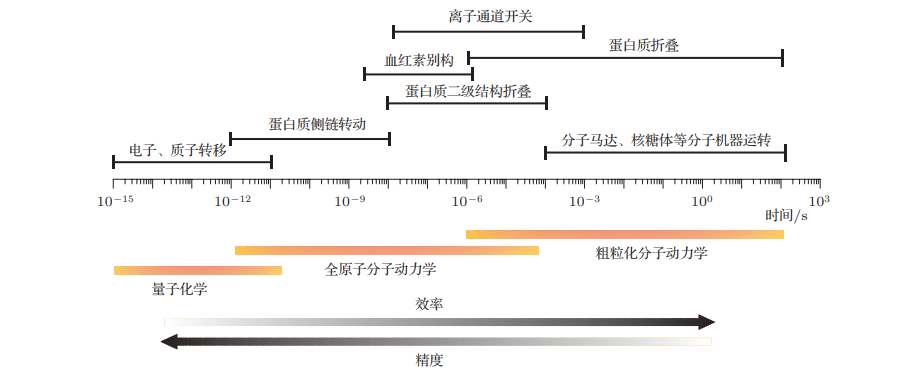
MD模拟基本思想很简单,即通过给定生物分子系统中所有原子的位置(例如图1左侧的蛋白质),就可以计算出每个原子受其他所有原子(除自身)的力。以图1左侧标注的红色氧原子为例,我们可以利用牛顿运动第二定律来预测氧原子的空间位置随时间的变化。在设定的时间中一步接着一步,反复计算氧原子受其他原子的力,然后利用这些力和位置坐标来计算氧原子新的位置和速度。可想而知,该过程的所需的计算量极大,要计算所有原子的空间位置的变化。所以说该过程所产生的轨迹本质上是一个三维电影,描述了在模拟时间间隔内系统原子级的构造。 图1 MD模拟基本思想流程图 那么模拟时间的间隔如何选取?一般来讲,为了保证数值的稳定性,MD模拟中的时间步长必须很短,通常只有几飞秒(10-15s)。大多数与生物化学相关的过程(图2),例如,蛋白质中功能重要的结构变化发生在纳秒、微秒或更长的时间尺度上;而蛋白质的侧链变化在皮秒的时间尺度上就可以发生。因此,一个典型的模拟涉及的时间步长多则数十亿。所以基于时间步长的基础上,并要在单个时间内评估数百万次原子间相互作用,导至模拟计算的要求非常高。 图 2 典型生物大分子动力学的时间尺度以及常用模拟计算方法 二、MD是怎样的原理?
网上有很多关于MD的理论计算原理的介绍,对没有数学/物理基础,或是这方面较为薄弱的人而言,理解该原理是比较困难的。所以这里我们尽量通过少的公式,形象的图片,来介绍该过程的原理。
牛顿力学 从刚刚的MD基础,可以得知整个分子动力学模拟是基于经典力学的,也就是要满足公式(1)。从公式可以看出,若想计算某一原子经过Δt时间的坐标位置变化,就需要知道该原子在系统中受到的所有力。那么这个力在分子动力学模拟当中如何得到呢?实际上通过公式可以发现,就是通过系统的所有原子的势能而得到的。
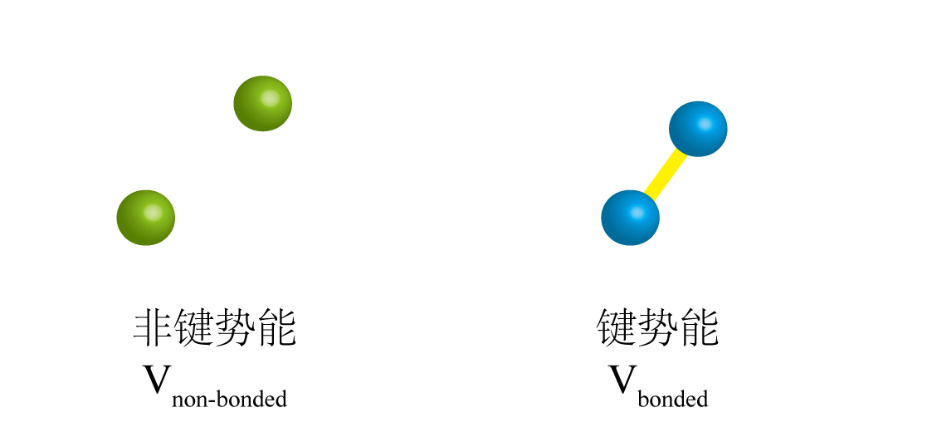
势能 那么势能该如何求得?对于不同力场有不同组成,这里仅介绍几个基本元素。对于一个复杂的分子体系,应包含键的势能和非键的势能,如式(2)和图3。也就是说,相距较远的原子之间是没有键的约束,那么彼此之间的势能是通过原子间相互作用力描述的;相反,相距较近的原子是通过键联系在一起的,是利用偏离平衡态的程度来描述原子的势能。
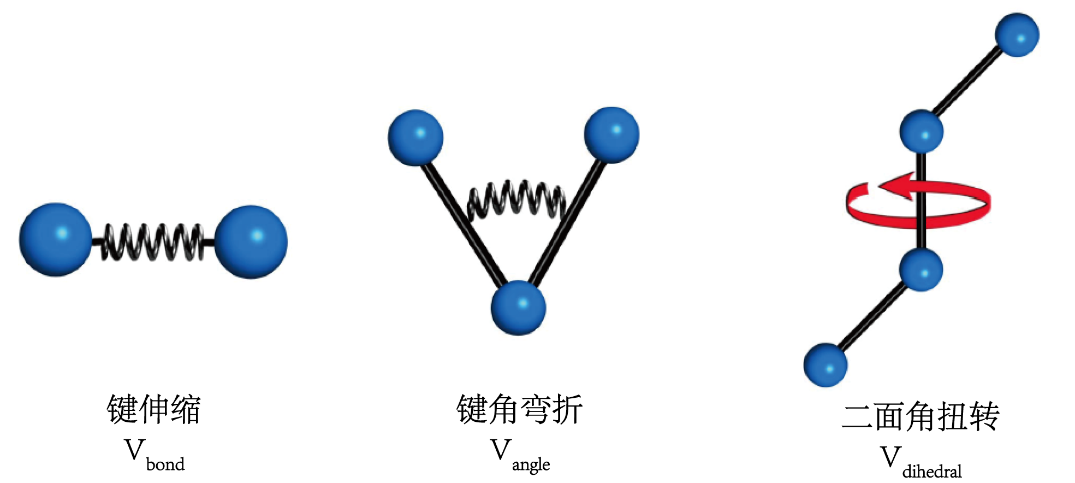
图 3 非键势能和键势能 非键势能包含两项,即:范德华相互作用(Vvdw)和静电相互作用(Vele)(式3)。所谓的范德华相互作用是存在分子间的一种相互作用,主要包含吸引力和排斥力;静电相互作用是由于原子吸引电子的能力不同,导至空间上的静电势不均匀分布而产生的。从图4可以看出,范德华相互作用和静电相互作用都是和原子之间的距离相关的。范德华力属于短程力,力的作用范围很小,影响力随距离的增加而急速减小,达到一定距离后作用力消失;而静电力是一种长程力,作用强度随距离的增加而缓慢减少。 图4 非键相互作用 键的势能包括:键长势能(Vbond)、键角势能(Vangle)、二面角势能(Vdihedral)(式4)。键长势能的变化主要是由于两个原子之间键会发生伸缩(图5);键角势能是由于连接三个原子之间的两个键形成的角发生改变(图5);二面角势能会发生改变的原因是:在四个原子组成的三个连续的键中,位于中间的键发生旋转,其他两个键不发生转动,旋转键与另外两个键分别形成两个平面,那么这两个平面的二面角就会发生变化(图5)。
图5 键的相互作用项
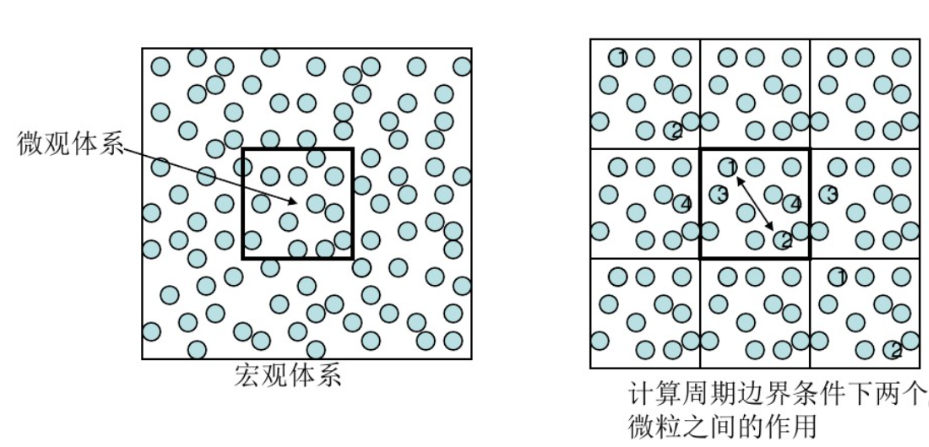
边界条件 分子动力学模拟实质上是想通过描述微观下的状态来得到宏观性质,这就需要有足够的粒子,但这无疑是加大了计算量。所以为了减小计算规模,研究人员引入了边界条件,在分子动力学模拟中常用到的边界条件就是周期性边界条件。选用周期性边界条件的原因主要是,在粒子的运动过程中,若有一个或几个粒子跑出模型,则必有一个或几个粒子从相反的界面回到模型中,从而保证该模拟系统的粒子数恒定;并且对于处于边界处的原子受力全面,无边界效应(图6)。 图6 周期性边界条件 对原理公式及推导过程感兴趣的朋友们,可在文章末尾去查看相关链接。在下篇中,我们会介绍分子动力学的准确性和步骤流程,也欢迎小伙伴们和我们讨论CADD的相关内容!
MD模拟相关原理公式以其推导过程 Tuckerman, M.E., Martyna, G.J.: Understanding modern molecular dynamics:Techniques and applications, J.Phys. Chem. B. 104, 159–178 (2000) https://www.slideshare.net/HamedHoorijani/molecular-dynamics-review
参考文献 [1]. Hollingsworth SA, Dror RO. Molecular Dynamics Simulation for All[J]. Neuron. 2018 Sep 19;99(6): 1129-1143. doi: 10.1016/j.neuron.2018.08.011. [2]. 李文飞, 张建, 王骏, 王炜. 生物大分子多尺度理论和计算方法[J]. 物理学报, 2015, 64(9): 098701. doi: 10.7498/aps.64.098701. [3]. Kumar Hemant, Maiti Prabal. Introduction to Molecular Dynamics Simulation[J]. 2011. 10.1007/978-93-86279-50-7_6. [4]. 陈正隆.分子动力学模拟与实践[M]. 化学工业出版社, 2007. [5]. Din XueDong, Michaelides Efstathios. Kinetic theory and molecular dynamics simulations of microscopic flows[J]. Physics of Fluids. 1997, 9. 10.1063/1.869490.
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2022-11-14 11:49
发表于 2022-11-14 11:49
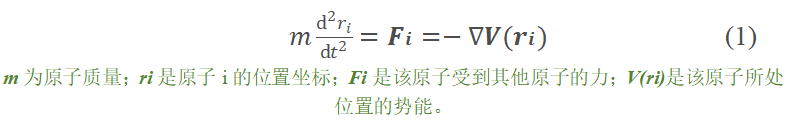
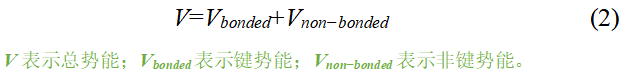

 提升卡
提升卡


