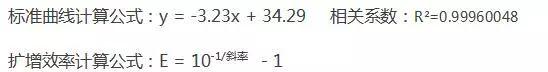用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
PCR技术
›
万字长文讲清荧光定量PCR
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
6524
|
回复:
0
[分享]
万字长文讲清荧光定量PCR
[复制链接]
空白派
空白派
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2025-5-10 05:31
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
刚接触到荧光定量PCR的时候,面对一堆名词难免抓瞎。
扩增曲线、标准曲线、域值、CT值、熔解曲线、基线到底都是什么意思?
那些荧光图色彩斑斓的很漂亮但是我好像不认识它?
今天我就从用万字长文,从荧光定量PCR的入门讲到高阶。
<hr/>
1.基本认识
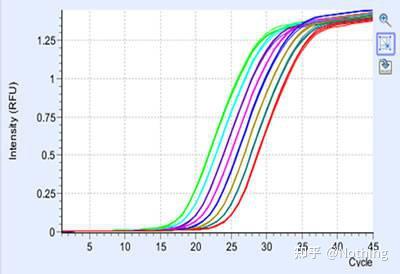
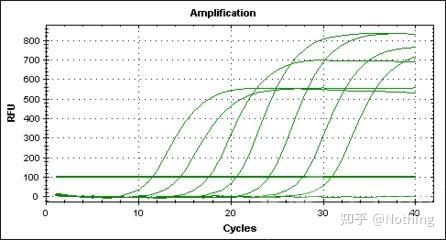
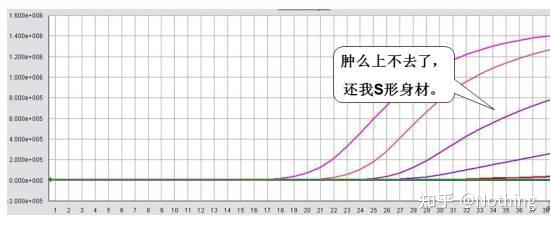
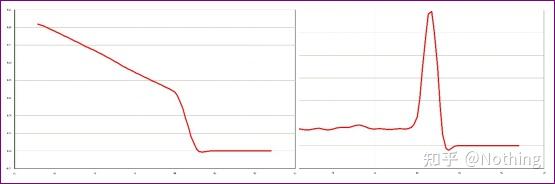
扩增曲线
扩增曲线
是指PCR过程中,以
循环数为横坐标
,以反应过程中实时
荧光强度为纵坐标
所做的曲线。
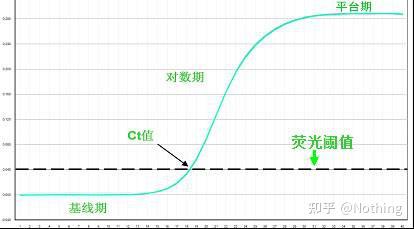
基线(Baseline)
基线
是指在PCR扩增反应的最初数个循环里,荧光信号变化不大。接近一条直线,这样的直线即是基线。
荧光域值(threshold)的设定
一般将 PCR反应
前 15个循环
的荧光信号作为荧光本底信号,
荧光域值
是PCR3—15个循环荧光信号标准差的10倍,荧光域值设定在 PCR扩增的指数期。一般来说,每台仪器在使用前都已经设置好了荧光阈值。
CT值
CT值表示每个PCR反应管内荧光信号到达设定的域值时所经历的循环数
。研究表明,各模板的CT值与该模板的起始拷贝数的对数存在线性关系,
起始拷贝数越多,CT值越小,反之亦然
。利用已知起始拷贝数的标准品可作出标难曲线,其中横坐标代表起始拷贝数的对数。纵坐标代表CT值。因此,只要获得未知样品的CT值,即可从标准曲线上计算出该样品的起始拷贝数。
判断扩增曲线是否良好的指标主要有几个方面:
A:曲线拐点清楚,特别是低浓度样本指数期明显,扩增曲线整体平行性好,基线平而无上扬现象,低浓度样本扩增曲线指数期明显。
B:曲线指数期斜率与扩增效率成正比,斜率越大扩增效率越高。
C:标准的基线平直或略微下降,无明显的上扬趋势。
D:各管的扩增曲线平行性好,表明各反应管的扩增效率相近。
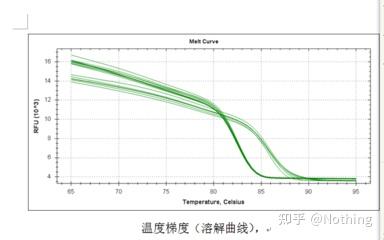
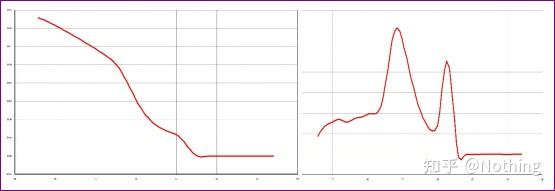
熔解曲线
对PCR产物加热,随着温度的升高,双链扩增产物逐渐解链,导致荧光强度下降,到达某一温度时(Tm),会导致大量的产物解链,荧光急剧下降。利用该特点以及不同PCR产物其Tm值的不同,因此使其荧光信号发生迅速下降的温度也不同,可通过此对PCR的特异性进行鉴定。
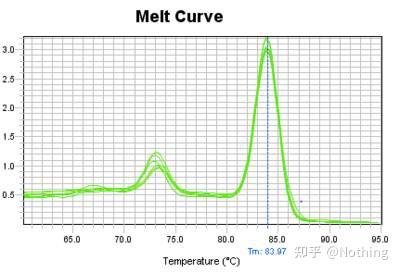
熔解曲线(取对数曲线)
对熔解曲线取对数,形成峰图,更直观的显示产物片段的情况。由于熔解温度即是该DNA片段的Tm值,以此可以判断影响DNA片段Tm值的一些参数,比如片段大小、GC含量等等。一般来说,根据我们的引物设计原则,
扩增产物长度在80-300bp范围,那么熔解温度应该是在80℃-90℃之间。
A:如果在80℃-90℃之间出现唯一主峰,说明荧光定量PCR完美;
B:如果在80℃-90℃之间出现主峰,80℃以下出现杂峰,基本考虑引物二聚体,可尝试提高退火温度解决;
C:如果在80℃-90℃之间出现主峰,温度上升又出现杂峰,基本上考虑有DNA污染,需要在实验初始阶段去除DNA。
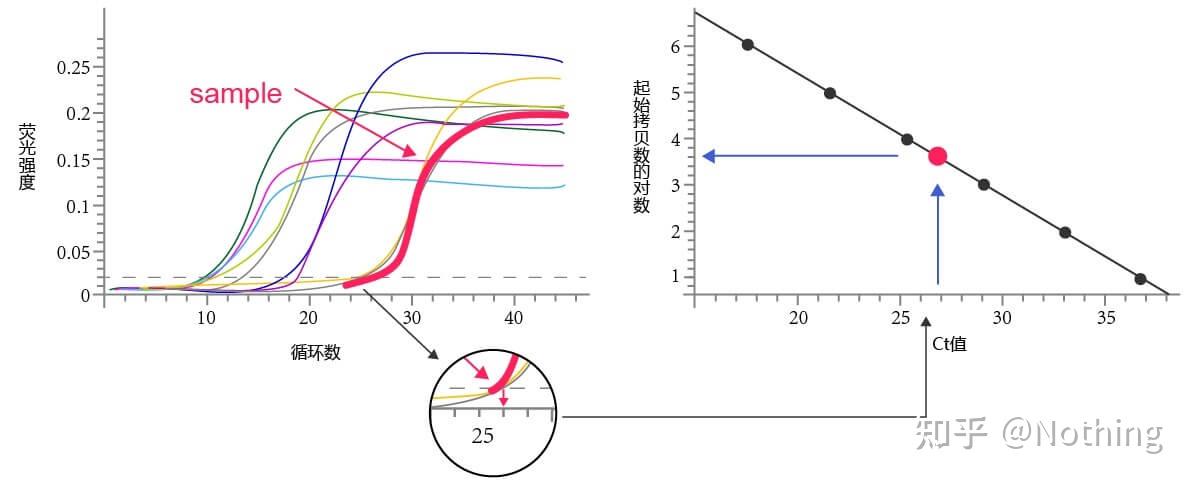
标准曲线
将标准品稀释至不同浓度,作为模板进行PCR反应。以标准品拷贝数的对数值为横坐标,以测得的CT值作为纵坐标,绘制标准曲线。对未知样品进行定量时,根据未知样本的CT值,即可在标准曲线中得到样品的拷贝数。标准曲线在绝对定量的时候非常重要。
Q:RT-PCR、QPCR、Real-time PCR、real-time RT-PCR有什么区别?
A:
RT-PCR就是逆转录 PCR
(reverse transcription PCR,RT-PCR),是聚合酶链式反应(PCR)的一种广泛应用的变形。在RT-PCR中,一条RNA链被逆转录成为互补DNA,再以此为模板通过PCR进行 DNA扩增。
Real-time-PCR
和
qPCR
(Quantitative Rea-ltime-PCR )是一码事,都是实时定量PCR,指的是PCR过程中每个循环都有数据的实时记录,因此可以对起始模板数量进行精确的分析。
虽然Real-time PCR(实时荧光定量PCR)和 Reverse transcription PCR(反转录PCR)看起来都可以缩写为RT-PCR,但是,国际上的约定俗成的是:
RT-PCR特指反转录PCR
,
而Real-time PCR一般缩写为 qPCR(quantitative real-time PCR)。
而real-time RT-PCR(RT-qPCR),
就是结合了荧光定量技术的反转录PCR
:先从 RNA 反转录得到 cDNA(RT),然后再用 Real-time PCR进行定量分析(qPCR)。
Q:为什么荧光定量PCR的扩增产物片段长度应该控制在80-300bp范围内?
A:每个基因序列长短不一,有的好几kb,有的几百bp,但是我们在设计引物的时候只需要要求产物长度80-300bp,太短或者太长都不适合做荧光定量PCR检测。
产物片段太短,无法跟引物二聚体区分,引物二聚体的长度大概在30-40bp,小于80bp很难区分是引物二聚体还是产物。产物片段太长,超过300bp,容易致使扩增效率低下,不能有效的检测出该基因的量。
打个比方,你在数一间教室里面有多少个人 的时候,只需要数一下有几张嘴就可以了,在检测基因的时候也是一样的,只需要检测某个基因的某一段序列来代表整个序列即可。如果你为了想数多少个人,既要数多少张嘴也要数多少个鼻子、耳朵、眼镜,反而容易数错。
Q:引物设计最佳长度是多少?
A:一般来说,引物长度大概在20-24bp范围是比较好的。当然我们在设计引物的时候一定要注意引物的TM值,因为这个关系到最佳退火温度。经过大量的实验证明,60℃是一个比较好的TM值。退火温度太低容易导致非特异性扩增,退火温度太高一般会导致扩增效率比较低,扩增曲线起峰比较晚,CT值延后。
Q:采集样本量的多少会不会影响实验结果?
A:不会。很明显,采集样本量多,提取的RNA就比较多,cDNA就比较多, 目的片段就比较多。对于绝对定量,要计算出某个目的片段的拷贝数,那么采集样本量的多少肯定会影响实验结果。比如检测血液里面的乙肝病毒HBV,就是检测出一定量(1ml)血液里面的HBV含量。
对于科研工作中常用的相对定量,样本量的多少与实验结果无关,因为相对定量是指目的基因和内参基因相比较,你可以认为它们是存在于同一核酸链条的上下游片段,如果样本量多了,内参基因和目的基因都同时等比例增加,不影响结果。
Q:反转录酶效率高低会不会影响实验结果?
A:同上。此处必须注意,我们希望获得较高的反转录效率,更希望反转录酶的反转录效率比较稳定,得到均一性的结果。这就要考验各大公司对于反转录试剂盒的优化能力了。
Q:Taq酶的效率会不会影响实验结果?
A:Taq酶的效率影响比较大,一般要求用热启动Taq酶,且效率要比较高才行,商品化的荧光定量试剂盒,每个厂家都会在自己的产品的基础上,把效率优化到最好的状态,接近于100%,效率太低实验结果不可用。对于不同公司的产品,这是衡量优劣的指标。
Q:荧光染料的多少会不会影响实验结果?
A:当然是有影响的。荧光染料过于饱和,会导致某些仪器出现噪点干扰;荧光染料不饱和,荧光值太低,导致提前进入平台期,扩增曲线平缓。在荧光定量实验中主要看CT值,因此后期扩增曲线进入平台期显得不那么重要,只是图形不够美观罢了,如果二者必选其一的话,宁愿选择荧光染料略微不饱和。在使用同一公司的同一款产品时,该影响基本可以忽略。
Q:PCR管的透光度会不会影响实验结果?
A:当然是有影响的。但是对于同一厂家同一批耗材,这种影响可以忽略。推荐大家用96孔板加高透膜,使耗材的透光度影响降到最低。
Q:操作过程中的误差会不会影响?
A:操作过程的影响,主要体现在均一性上。均一性是指体系内的所有组分均匀的混合在一起,瞬时离心可以解决均一性问题。另外,对于生手来说,最好调整PCR体系在20ul以上,过小的体系更容易产生误差。再请看下图:如果退火温度优化得合适,引物浓度对CT的影响会降到最低。 你会明白,有的操作误差(比如引物浓度)是可以通过优化退火温度来避免的。
<hr/>
2.进阶认识
关于实时荧光定量PCR,我们必须了解这样一个现实,每年发表的成千上万的科研论文,其中利用到荧光定量PCR技术的不是小数目。
那如果没有一个共同的标准来衡量荧光定量PCR实验,结果可能千差万别,对同样的物种同样的基因,用同样的处理方式,检测出来的结果也是千差万别,后来者也难以重复出同样的结果。
这是不是说荧光定量PCR就是一个骗子技术或者说是一个不靠谱的技术?
并不是,就是因为荧光定量PCR比较敏感比较精确,导致一点错误的操作就会出现完全相反的结果。
失之毫厘谬以千里
。文章作者可能会被审稿人反复折磨,同时,杂志审稿人也被不同的实验结果难以取舍。
所有的一切,都指向荧光定量PCR实验没有达成共识。
为此,行业资深科学家开始制定标准,
要求投稿人在文章中提供一些必要的实验和数据处理的细节(包括必要的数据)来满足这些标准
。
审稿人读到这些细节就可以判实验的质量;将来的读者也可以据此来重复试验或者改进实验。那么这样出来的实验结果就是充满了信息量、是高质量的,是可用的。
MIBBI(Minimum Information for Biological and Biomedical Investigations -
http://www.mibbi.org
) 应运而生。
MIBBI是一个为实验提供标准的项目
,发表在nature上,这个项目针对各类生物学试验,包括细胞生物学、Microarray、我们现在要讨论的qPCR等等,给每一类实验规定了在提交稿件时是都应该提供那些信息。
MIBBI项目中,与荧光定量PCR相关的文章有两篇,分别是:
RDML (Real-Time PCR Data Markup Language )——实时定量PCR数据的结构化语言和报告指南;
MIQE(Minimum Information for Publication of Quantitative Real-Time PCR Experiments)——用于发表关于实时定量PCR实验文章的最小信息。
今天,我讨论的重点是RDML,也就是术语规范。
凡事如果没有标准定义,就无法继续讨论,这也是为什么名词解释在考试中如此重要的原因。
荧光定量PCR实验用到的术语包括以下内容,QIAGEN公司已经为我们做了最佳的总结,以下全是干货,
建议学霸收藏不定期食用
。
基 线
基线是
早期循环的噪点水平
,通常在第3和第15循环之间测量,这是因为在此期间还检测不到扩增产物引起的荧光值增加。用于计算基线的循环数量是可以改变的,如果使用高模板量或者靶标基因的表达水平高则循环数量需要减少。
设置基线需要查看线性度扩增曲线的荧光数据
。设置基线,使得扩增曲线的增长所开始的循环圈数大于基线循环最高圈数。对每个靶标序列都需要单独设置基线。早期循环检测到的平均荧光值需要从扩增产物中获得的荧光值中减去。各种Real-Time PCR软件的最新版本允许单个样品自动优化基线设置。
本 底
本底是指
反应中的非特异性荧光值
。例如:低效率的荧光淬灭;或由于使用SYBR Green造成的大量双链DNA模板。信号的本底分量由Real-Time PCR软件算法数学除去。
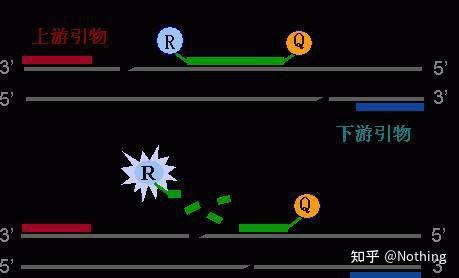
报告基因信号
报告基因信号是指在Real-Time PCR过程中由SYBR Green或荧光标记的序列特异性探针产生的荧光信号。
归一化报告信号(RN)
RN指的是报告染料荧光强度除以在每个循环中测量的被动参照染料的荧光强度。
被动参比染料
在某些Real-Time PCR中,
荧光染料ROX被用作荧光信号标准化内部参照
。它可以逐孔校正因移液不准确、孔位置及荧光波动造成的变动。
阈 值
阈值调整到本底值之上并显著低于扩增曲线的平稳值。它必须处于扩增曲线的线性区域内,代表了PCR检出的对数线性范围。阈值应在对数扩增曲线视图中进行设置,以使PCR的对数线性期易于识别。
如果Real-Time PCR中有多个目标基因,阈值必须为每个目标进行设定
。
循环阈值(CT)或交叉点(CP)
扩增曲线穿过阈值的循环(即,荧光检测值显著增加的点)。CT可以是一个分数,并且可以计算起始模板量。
ΔCT值
ΔCT值描述了
靶标基因和相应的内参基因CT值的差值
,例如看家基因,并用于标准化模板的使用量:
⇒ ΔCT = CT (靶标基因) – CT (内源性参比基因)
ΔΔCT值
ΔΔCT值描述了感兴趣样品(例如,刺激细胞)的平均ΔΔCT值与参考样本(例如,未刺激细胞)的平均ΔΔCT值之间的差值。参照样品也被称为校准样品,并且所有其它样品进行相对定量时都将被标准化到如此:
⇒ ΔΔCT = 平均ΔCT (感兴趣样品) – 平均ΔCT (参比样本)
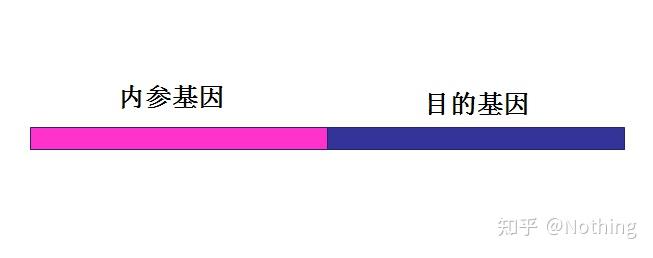
内源性参比基因
内源性参比基因的表达水平在样品间不存在差异
,例如看家基因。对比内参基因与靶标基因的CT值可以将靶标基因的表达水平归一化到输入的RNA或cDNA的量(见上面关于ΔCT值部分)。
内参基因对以下情况作出校正:
可能的RNA降解或RNA样品中存在酶抑制剂的情况,以及RNA含量、逆转录效率、核酸回收和样品处理的变动。为了选出最优的参照基因(多个),我们对算法进行了改进,允许其选择依赖于实验设置最优参照。
探针法荧光定量PCR原理
内 参
在同一反应中被作为靶序列扩增的,并用不同的探针(即,进行双重PCR)检测的对照序列。内参经常被用来排除失败的扩增,例如没有检测到目标序列的情况。
标定样品
在相对定量中使用的参比样品(例如,源自细胞株或组织纯化的RNA),用以与所有其他样品进行比较,以确定某一基因的相对表达水平。标定样品可以是任何样品,但通常是一个对照品(例如,未处理的样品或来自实验零时的样品)。
阳性对照
使用已知量模板的对照反应。阳性对照通常用来检查引物集或引物–探针集工作是否正常,以及反应是否正确设置。
无模板对照(NTC)
包含除模板之外的所有扩增反应所必要组分的对照反应。这使得发现由于污染的试剂或外源DNA造成的污染成为可能,例如从以前的PCR中。
无RT对照(NRT)
RNA制备物可能含有残留的基因组DNA,如果检测不被设计为仅检测和扩增RNA序列,其可以在Real-Time PCR中进行检测。DNA污染可以通过操作在其中没有加入逆转录酶的RT对照反应来进行检测。
标准品
标准品是指已知浓度或拷贝数,用于构建标准曲线的样品。
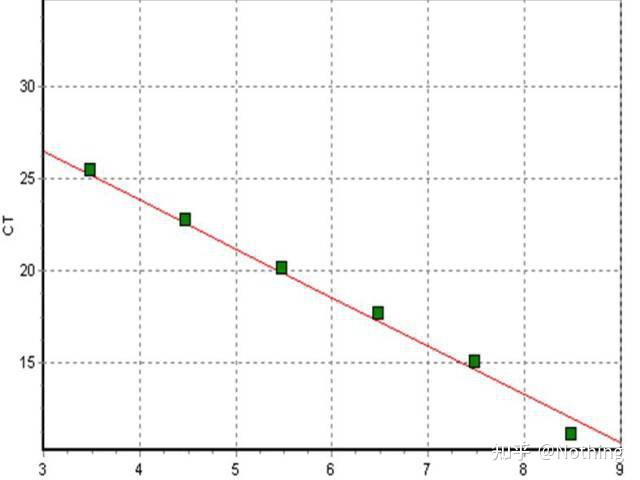
标准曲线
要生成标准曲线,
用CT值/不同标准稀释的交叉点对标准物质输入量的对数绘图
。标准曲线通常是使用至少5种不同浓度的标准稀释倍比生成。每个标准曲线,应检查其有效性,斜率值落在–3.3到–3.8之间。标准是各浓度一式三份的理想测定值。标准曲线的斜率如果与这些值差异较大则应该舍弃。
效率和斜率
标准曲线的斜率提供了对real-time PCR的效率指示。斜率 –3.322表示该PCR具有数值为1的效率,或100%的效率,并且PCR产物的量在每个循环增加到两倍。效率小于–3.322(例如,–3.8)则表示PCR的效率<1。
通常,大多数扩增反应因为实验的限制不能达到100%的效率。斜率大于–3.322(例如,–3.0)表明PCR效率似乎是大于100%的。当在反应中的非线性期对值进行测量时可能发生这种情况,或者它可以指示是否有抑制剂存在。
以上标准术语是不是似曾相识的感觉,是的,在基础版本里面也会看到,这里更加详细。
<hr/>
3.高阶认识
MIQE(
M
inimum
I
nformation for Publication of
Q
uantitative Real-Time PCR
E
xperiments)——
用于发表关于实时定量PCR实验文章的最小信息
。
要使自己的实验有说服力,不得不了解MIQE,为了简化大家的理解,我们将重点内容简化。
MIQE原文大家可以在网上搜索到,其中最重要的是其规定了
发表文章的时候需要提供的数据核查清单
,为了让大家速成学霸,大师兄将重点都给你总结了。
MIQE核查清单
本期MIQE的信息虽然比较多,但都是为我们的实验提供精准的信息。
审稿人读到这些细节就可以判实验的质量;将来的读者也可以据此来重复实验或者改进实验。
实验的首要原则,是要确定
实验逻辑的严密性
。最根本的是实验设计,而实验设计最重要的是如何设定目标样本、参考样本(对照)、重复数量,让实验数据具有参考性、可比性和说服力。
目标样本
是指经过某种处理后需要我们检测目的基因的样本。
参考样本
即没有做任何处理的样本,生物学上常常说的野生型。
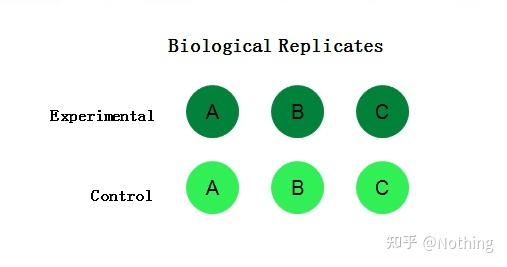
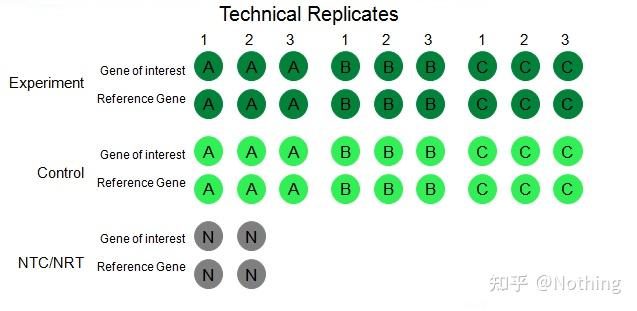
实验的重复
(Experimental Replicates)非常重要,具有说服力的重复数量一般要达到三个以上,要区分什么是生物性重复、什么是技术重复。
生物性重复
(Biological Replicates):不同的材料(时间、植株、批次、反应板)做的同一验证实验。(同一本王镜岩的《生物化学》虐几个学渣)。
我们以农药处理辣椒为例子,我们要对ABC三个植株进行农药喷洒,那么ABC三个植株就是三个生物性重复,它们就是不同的材料进行的同一验证实验。但是作为实验肯定是需要对照的,所以我们在操作的时候可以对A植株的其中一根枝条进行喷洒,形成A植株的实验组,对A植株的其他枝条不喷洒,形成对照组。对B和C也进行同样的处理。
技术重复
(Technical Replicates):是为了避免操作上导致的误差而设计的重复实验,实际上就是同一材料所包括的复孔。(用王镜岩的《生物化学》虐几个学渣N次,然后取平均数)。处理和对照都要有目的基因和内参基因的重复设置(最少三个)。
再拿农药处理辣椒作为例子,对于A植株实验组,我们分别对它的目的基因和内参基因做1、2、3三个PCR复孔,以待检测完以后取平均数,对于A植株的对照组也作同样处理。同样,对于B、C植株也作同样的处理。这就是技术性重复。
值得注意的是:进入统计的是生物性重复,而技术重复是检验实验过程中是否有任何随机现象使实验结果可信,也就是为了我们经常说的取它们的平均数避免误差。
阴性对照
,又分为NTC与NRT。
NTC
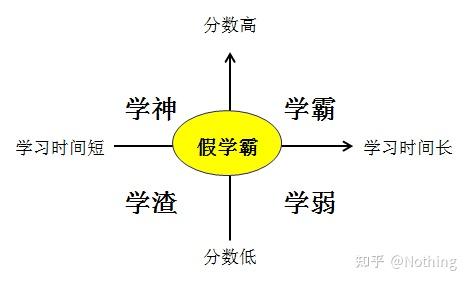
(No-Template Control),没有模板的对照,用来验证实验材料是否具有污染。一般以水作为模板,如果有荧光反应,说明实验室有核酸污染发生。
这些污染来自于:水不纯,不合格的试剂含有内源DNA,引物污染,实验室器材污染,气溶胶污染等等,需要使用RNase清除剂和RNase抑制剂。气溶胶污染是最不容易发现的,想象一下你的实验室就像雾霾,空气中悬浮着各种核酸。
NRT
(No-Reverse Transcriptase),没有反转录的对照,是未经反转录的RNA作为阴性对照,这是对gDNA残留的控制。
在做基因表达时,通过检测反转录后的cDNA量来检测RNA的量,那么如果在提纯RNA时有gDNA的残留,就会造成实验结果的误差,因为实际得到的结果是gDNA和cDNA的总体水平而不仅仅是cDNA的,需要在提取RNA的时候彻底去除gDNA。
有的学霸自作聪明,费尽心思的设计跨内含子引物来避免gDNA,我想说他可能是个假学霸,我们以后会讨论这个问题。
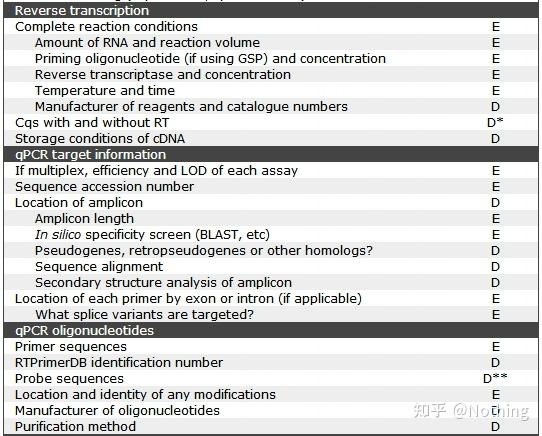
MIQE之样本信息
我们在发表关于qPCR的文章的时候,一定要讲明白样本信息,这是文章不可或缺的部分。同样,我们在处理样本的时候,也要同样规范自己的操作,以保证样本的有效性。
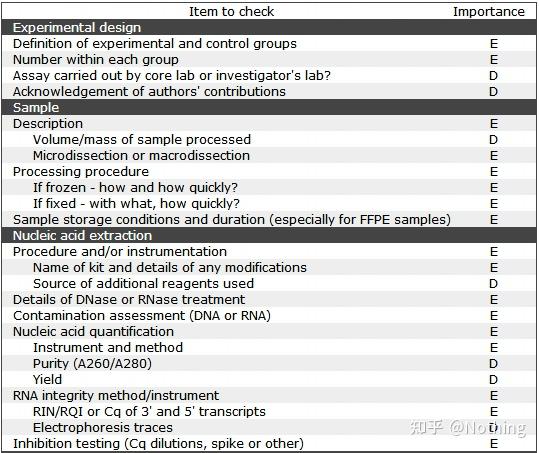
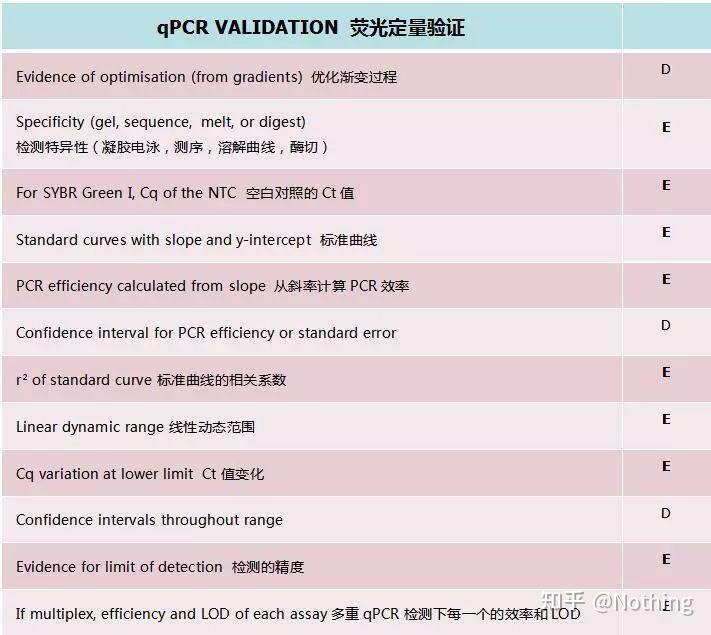
在MIQE核查清单里,有D和E两个字母表示该项的重要性,
E:essential information基本信息(必须提交);
D:desirable information理想的信息(尽可能提供)。
下面这个表格就是核查清单里样本信息要求:
对样本的描述只是一个结果,而我们更应该注意的是整个实验过程中的取材。
实验材料的选择
血液样本——选择新鲜血液,不得超过4小时。
细胞样本——选择处于生长旺盛的时期收集新鲜的细胞。
动物组织——选择新鲜、生长旺盛的组织。
植物组织——选择新鲜的幼嫩组织。
学渣们一定注意到这几句话中都有一个关键词:新鲜,所以说小鲜肉并不是大妈才喜欢,实验室的师姐师妹都喜欢。
实验材料的保存
一般来说,我们不建议对样本进行保存,如果条件允许的话。但是不乏有很多朋友无法在采样后立刻进行实验,有的甚至需要背着液氮罐到野外采样。
对于这种苦逼朋友,我只能说你不了解试剂耗材,现在很多试剂耗材公司生产出可以常温保存RNA样本的试剂,可以选择使用。因为他们都没有给广告费,所以这里就不介绍哪些厂家的产品了。
常规的保存方法就是液氮保存。
把样本带回实验室以后,保存在-80℃冰箱即可。
值得注意的是:对于牵涉到RNA的实验,六字原则必须遵守:
低温
、
无酶
、
快速
。
低温
,这个概念容易理解。
无酶
,我们生活的世界到处都是RNase(要不然你早就被HIV搞死了),所以在做实验的时候如何避免RNase,是一种很重要的观念。
快速
,天下功夫无坚不破,唯快不破。
有的女孩子做实验很仔细,但是做几年也不如一个灌篮高手做得好,觉得上天不公平,怨天尤人,寻死觅活,其实她没有明白,就是因为她太仔细做事情很慢,反而没有保护好RNA,而灌篮高手手脚灵便,做实验的时候还想着三下五除二搞定了灌篮去,反而把实验做好了。
注意:
慢了,RNase入侵的机会就更多了。怎么训练自己快速呢?没有办法,多练习。
对于样本的取材和保存过程,MIQE都要求必须清楚明白的写在论文中,以方便审稿人审阅论文的可靠性,同时也方便后来的愣头青们重复你的实验。
生物学实验虽然难但是高端,一不小心可以颠覆世界,比如搞个SARS变成生化危机,比如搞个杂交水稻拯救13亿人口。
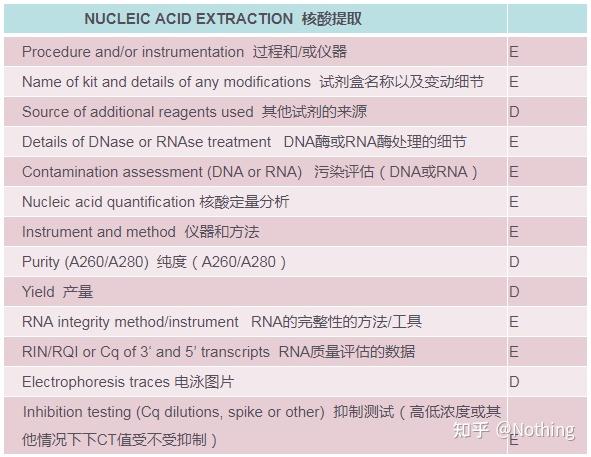
MIQE之核酸提取
核酸提取是一个大事,所有的分子生物学实验都是从核酸提取开始。首先,我们还是照抄一下MIQE关于核酸提取的内容。
再次强调,在MIQE核查清单里,有D和E两个字母表示该项的重要性。
E:essential information基本信息(必须提交);
D:desirable information理想的信息(尽可能提供)。
看这个表格不能停留在表面,表格就是教条,要做学霸一定要多问个为什么。
这个表格的本质内容是:追求RNA的纯度、完整性、一致性、提取量。
接下来就一一分析这个表格是如何通过条款来追求这些的。在涉及到荧光定量PCR的文章中,需要对以上表格里面的项目作详细描述。
过程或者仪器
第一个部分就是核酸提取的步骤,如果用核酸自动提取仪提取(高级哦),需要注明仪器的型号名称。
这台北京济凡的96通道的核酸自动提取仪摆在这里,你看不清logo应该不算打广告。
试剂盒名称以及变动细节
用的什么试剂盒,加了什么特殊试剂或者做了什么特殊操作要讲明白,以便别人能够方便的重复出你的实验。
有的人在提取特殊样本的时候加了一些特殊试剂,认为这是自己的秘密武器不告诉别人,保密的同时,也失去了让你的文章大放光彩的机会。千万别耍小聪明,做科研要做到比乡下老张还老实,你想耍小聪明,文章就会让你出笨笨。
记住,你们在订购试剂盒以及写入文章的时候
一定要记住试剂盒的产品编号
,试剂盒上一般有两个编号:Cat——产品目录号(产品编号、货号),Lot——产品批号(用于标明产品出于哪个批次)。看不懂的看看下面这张偷来的图片就明白了。
另外订购生物化学类试剂的时候经常会用到
CAS
号,我就一起普及了,CAS号是美国化学会专门负责给予每一个新出的化学药品的编号,一般是三个数字用短杠连接。如水的CAS号:7732-18-5。化学药品常常会有多个别名,但是CAS号是唯一的。订购药品的时候可以先查查它的CAS号。注意和
Cat
号进行区分。
言归正传,为什么要清楚明白的描述这些,其实也是为了在RNA提取质量上把关,仪器和试剂盒信息的加入,会让RNA提取一致性更好。
DNA酶或RNA酶处理的细节
荧光定量PCR很重要的问题是要防止DNA污染,有污染不实验。因此,必须要说明你用于处理DNA的过程,这是为了证明实验过程中的DNA保证已经得到完全彻底的去除。同样,如果是做DNA的定量,也要保证RNA被完全的去除。
一般来说,去除DNA的方法是提取RNA后用DNase处理。不过这都是比较老的方法了,商品化的RNA提取试剂盒,已经能够在提取过程中去除DNA。
注意
提取RNA的过程中去除DNA是非常危险的双刃剑,这会使得提取RNA的操作时间延长,增加RNA降解的风险,基本上就是在RNA得率和纯度之间做取舍。
另外,在硅基质吸附柱上加入的DNase量非常小,必须要优质的DNase才能达到效果,未经优化的DNase是无法做到快速彻底消化的,这一点非常考验商家技术水平。
当然,更有奇葩的商家吹嘘说不用DNase也能去除DNA,可以这么说,凡是吹牛说不用DNase就能彻底清除DNA的都是耍流氓。DNA是比较稳定的双链结构,不是吹吹风就灰飞烟灭的。
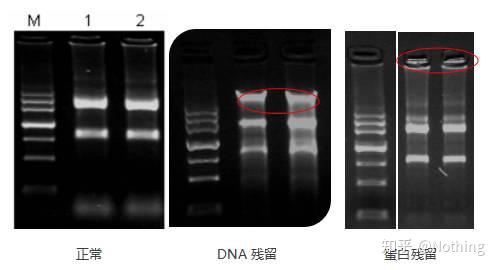
污染评估(DNA或RNA)
评估方法:电泳检测 1%琼脂糖,6V/cm,15min,上样1~3 ul。
核酸定量分析
通常使用紫外分光光度计进行测量 ,先给大家普及一下OD260、OD280、OD230三个值的含义。
OD260nm:是核酸最高吸收峰的吸收波长,最佳测量值的范围为0.1至1.0。如果不在此范围,稀释或浓缩样品,使之在此范围内。
OD280nm:是蛋白和酚类物质最高吸收峰的吸收波长。
OD230nm:是碳水化合物最高吸收峰的吸收波长。
接下来说各个指标的作用。对于A260来说,可以用来测定核酸的产量。当OD260=1时,dsDNA=50μg/ml,ssDNA=37μg/ml,RNA=40μg/ml。
对于纯度来说,就需要看那几个我们常见的比值了:OD260/280及OD260/230。
纯DNA:OD260/280约等于1.8,大于1.9时提示有RNA污染,小于1.6时提示有蛋白质及酚类污染。
纯RNA:1.7<OD260/280<2.0,小于1.7时提示有蛋白质及酚类污染,大于2.0时提示有异硫氰酸残留。
OD260/230:不伦是DNA还是RNA,参考值都为2.5,小于2.0时提示有糖类、盐类、有机物的污染。
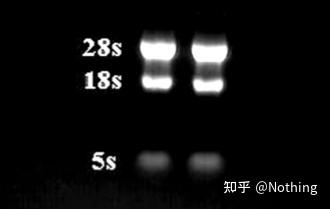
RNA的完整性的方法/工具
RNA的完整性非常重要,一般需要做一个RNA变性胶实验,检验28S和18S RNA之间的亮度是不是二倍关系。当第三条带5S出现的时候,说明RNA已经开始降解了,无脊椎动物除外噢。
RNA质量评估的数据:除了以上检测,在RNA完整性方面,还有一些比较高级的仪器检测,比如Experion全自动电泳系统RQI完整性检测,可以检测出RNA是否隐形的被降解。
之前在文章中已经说到,定量其实是目标基因和内参基因的比较,RNA的完整性是如何影响目标基因和内参基因之间的平衡,可以从下图简单的理解。降解会导致基因残缺,无论是内参基因的残缺还是目标基因的残缺,对数据都是有极大的影响。
抑制测试(高低浓度或其他情况下CT值受不受抑制)
以该图为例,五条曲线的Ct值分别如下。曲线之间CT值分布不均,高低浓度下Ct值均有延迟,这就是PCR受抑制的情况。
重点:在RNA提取过程中,我们需需要摒弃错误观念,树立正确观念。
错误的观念是:RNA提取只追求得率,认为获得的RNA量越大越好,实际上我们做定量的时候,如果基因数量不是很大,是要不了多少RNA的,你所提取的RNA量绰绰有余。
正确的观念是:RNA提取要追求纯度、完整性、一致性。纯度能够保证后续的反转录不受抑制以及能够保证不被DNA影响数据。完整性能够保证目标序列和内参的平衡。一致性能够保证上样量稳定。
明白了吗?没有!请收藏本文,多看几遍。
MIQE之反转录
之前我们说过,观念很重要,有了反转录的正确观念,才能跟随灵魂做对实验。
反转录的错误观念:
追求较高的上样量 。
反转录的正确观念:
追求一致性(稳定性),不管RNA上样量如何,反转录的效率都保持一致,确保cDNA的差异能够真实反映 mRNA 的差异。
这个教条具体如何呈现呢?
请看下图和解释:
反转录过程中的变化
反转录与PCR过程的区别
首先,我们要理解反转录过程与PCR过程的区别,PCR经过多次升温退火延伸的过程,目的片段呈指数增长;而反转录没有这个过程,我们可以想象反转录其实就是一对一的复制过程,有多少条RNA就得到多少条cDNA。
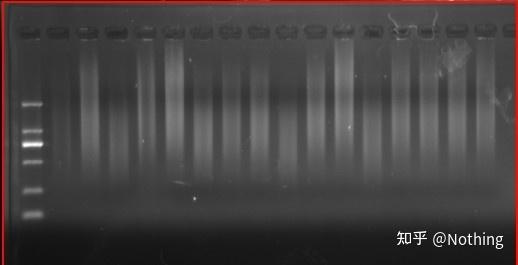
所以也告诉大家,反转录的结果拿去做电泳检测,基本上就是弥散的,如瀑布般,看不到任何有用信息,因为大大小小的片段都被反转录了,无法集中在一个片段上。由于RNA的量比较少,所获得的cDNA量也比较少,因此,基本上无法进行检测。
反转录后cDNA电泳结果
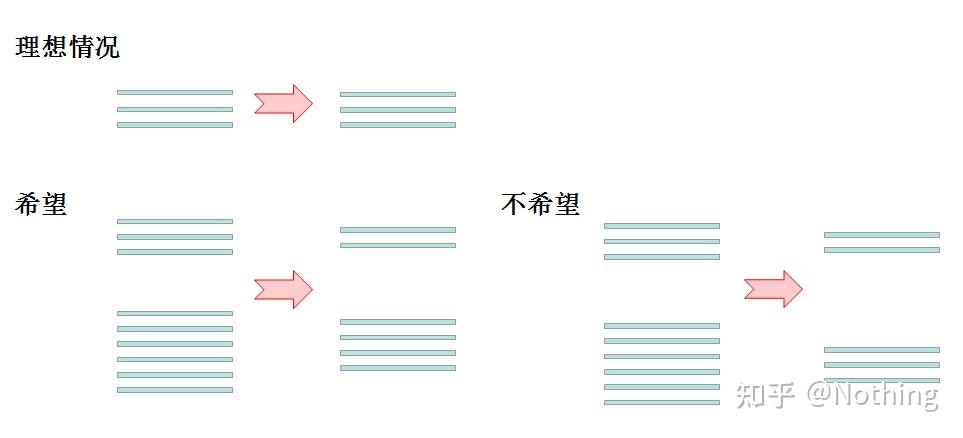
反转录稳定性
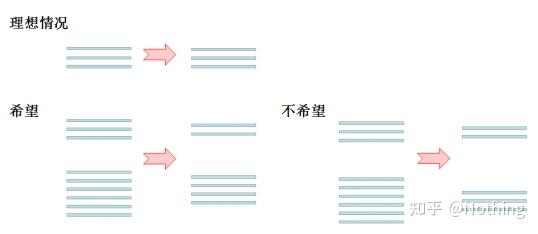
其次,解释上图反转录过程中的变化。理想情况下,反转录一对一进行,但是没有哪个公司的反转录酶能够达到这种效果,基本上大多数反转录酶的效率游走在30-50%之间。如果现实情况是这样的话,我们宁愿反转录效率能够比较稳定,也就是图中我们希望看到的部分:3条RNA得到2条cDNA,6条RNA得到4条cDNA,这样无论上样量多少,反转录效率都比较稳定。我们不希望看到反转录效率不稳定,高浓度被抑制的情况。
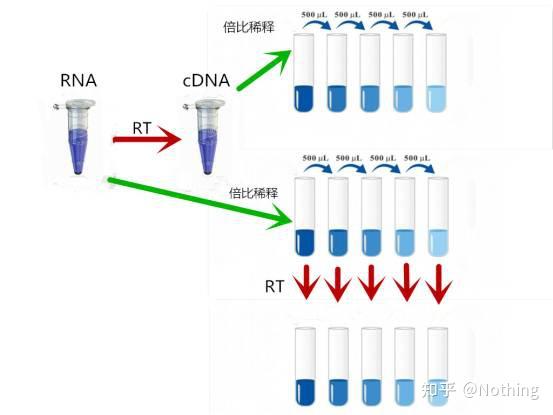
那么,如何验证反转录效率是否稳定呢?方法很简单,只需要做个对比试验:一是RNA进行倍比稀释后再反转录成cDNA,二是反转录成cDNA后再做倍比稀释,两者做qPCR,看看得到的斜率是否一致。
作为学霸的你应该秒懂。如下图:
检验反转录酶的效率是否稳定
反转录酶和试剂盒
完美的荧光定量PCR怎么少得了优秀的反转录酶以及试剂盒呢。所以,学霸们!再次敲黑板了!
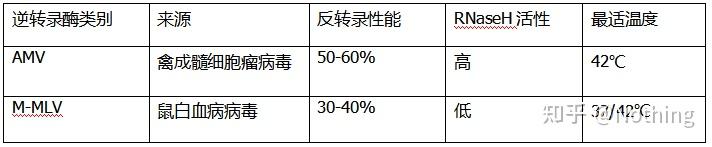
反转录酶根据来源大体上分两种,
AMV
或者
M-MLV
,他们的性能跟表格中显示的是一样一样的。
RNase H 活性
RNase H即Ribonuclease H,中文名为核糖核酸酶H,是一种核糖核酸内切酶,可以特异性地水解DNA-RNA杂合链中的RNA。RNase H不能水解单链或双链DNA或RNA中的磷酸二酯键,即不能消化单链或双链DNA或RNA。常用于cDNA第二链的合成。
这是一个奇怪的东西。我们说反转录酶有RNase H活性,并不是说反转录酶中含有RNase H,也无法从反转录酶中分离出RNase H,也许是反转录酶中某些基团的构象造成了该反转录酶有此活性存在。
所以,别看AMV反转录效率高一些,但是其RNase H活性使得cDNA的得率降低。当然,试剂厂商都在不断优化自己的产品,尽量消除反转录酶中的RNase H活性,提高cDNA的得率。
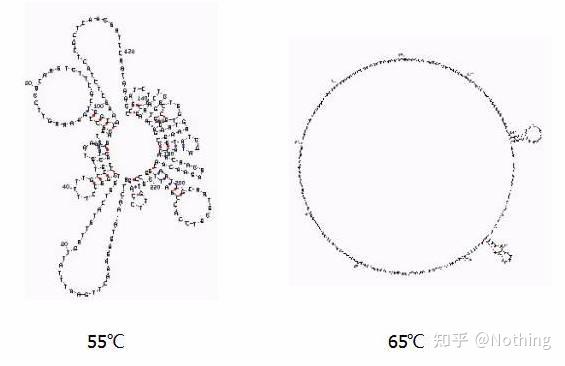
退火温度
不同温度下的二级结构
其次我们看看反应温度,看上图,用mFold在线工具,判断在特定温度和盐浓度条件下目标片段的二级结构。该RNA在55℃的情况下,二级结构还是非常复杂的,反转录酶无法工作,要到65℃才能完全解开二级结构,而AMV和M-MLV的最适温度都远远小于这个温度。
怎么办?
二级结构是模板本身自己互补配对,导致引物和反转录酶与模板结合面临很强烈的竞争,导致结果E很低、重复性很差,等等一系列的问题。
怎么办?
只有尽可能提高退火温度
。
很多试剂厂家都在改良自己的反转录酶,在笔者接触的产品中,
TIANGEN的Quant系列反转录酶
给出的解决方案是:通过基因克隆重组的方法,首先去除RNase H酶活性基团,其次是提高酶和RNA模板的亲和力。高亲和力可以竞争性地挤开二级结构,顺利的通读下去,同时也大大地提高了反转录效率。(保证没有收一分钱的广告费)
啥?其他公司有没有别的解决方案?笔者不知,他们也没有告诉过我。
划重点
反转录更重要的是在追求反转录效率的一致性(酶不仅要效率高还要稳定),而不是上样量,如果不是做特别大规模的荧光定量PCR,根本要不了那么多的cDNA。
各厂家在追求一致性的方面也做了些功夫,比如现在大部分公司都已经把反转录包装成为标准的试剂盒进行销售,这不失为一种很好的选择。
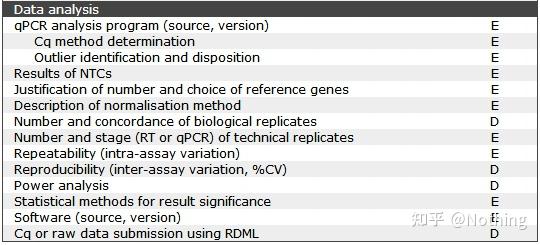
MIQE之目的基因信息
我们要讲的MIQE清单已经到了第五条。话说MIQE清单你还记得几式?你不会像张无忌那样一式也不记得了吧?
温故而知新,一共九条,列举如下(对待学渣要随时回味):
– Experimental Design (实验设计)
– Sample Information (样本信息 )
– Nucleic Acid Extraction (核酸提取)
– Reverse Transcription (反转录)
– qPCR Target Information(目的基因信息)
– qPCR Oligonucleotides(qPCR 寡核苷酸)
– qPCR Protocol (qPCR 程序)
– qPCR Validation (qPCR验证)
– Data Analysis (数据分析)
今天要说的是第五条 —— 目的基因信息。
(别慌,先上图后解释~)
E:essential information基本信息(必须提交);
D:desirable information理想的信息(尽可能提供)。
上图解释
1. 这个基因对于重复实验是否有效,一般通过重复实验可以得到验证。
2. 基因ID,你懂的。
3. 基因长度,目的基因总长度肯定没有问题的,在设计引物的时候,还要确保扩增子的长度在80-200bp之间,以保证有比较好的扩增效率。
4. 序列Blast比对信息,目的基因需要在genebank做比对,以防止非特异性扩增的出现。
5. 是否存在假基因。
假基因(pseudogene)与正常基因相似,但丧失正常功能的DNA序列,往往存在于真核生物的多基因家族中,常用ψ表示,是基因组中与编码基因序列非常相似的非功能性基因组 DNA 拷贝,一般情况都不被转录,且没有明确生理意义。
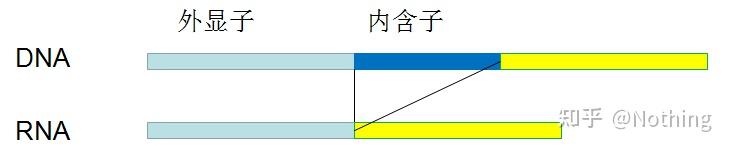
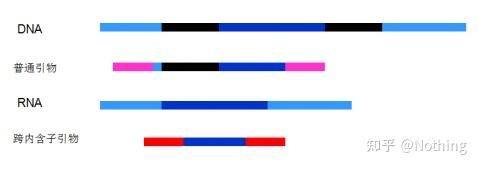
6. 引物相对于外显子和内含子的位置。
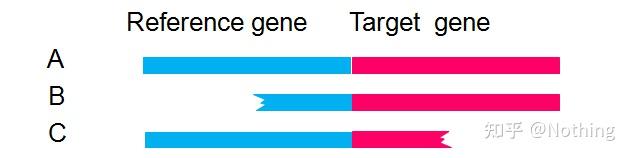
早年,我们在解决DNA污染问题的时候,常常会关注引物和外显子、内含子的位置,一般考虑跨内含子设计引物,避免将DNA扩增出来。
请看下图:黑色代表内含子,各种蓝色代表外显子,粉红代表普通引物,鲜红代表跨内含子引物。
这看起来是多么完美的计划~
而实际上,
跨内含子引物
在多数情况下没有想象中那么大的魔力,同样会引起非特异性扩增。所以为了防止DNA污染的最佳方法还是彻底去除DNA。
7. 构想预测
再次使用这个例子,用 mFold 在线工具,判断在特定温度和盐浓度条件下,目标片段的二级结构。
二级结构是模板本身自己互补配对 ,它会导致引物与模板配对面临很强烈的竞争,引物就机会少了,结果E很低、重复性很差,等等一系列的问题出现。通过软件预测,如果没有二级结构的问题,那再好不过了,如果有,我们后续的文章会专门讨论如何解决这个问题。
MIQE之qPCR 寡核苷酸,也就是引物
对于荧光定量PCR,你天天纠缠的第一件事是RNA提取,第二件事可能就是引物设计了。
首先,我们还是按照MIQE的核查清单来检验关于引物设计的规则,简单得让学渣们偷笑,一句话都可以讲完:搞清引物探针的序列和位置以及修饰方法。对于引物纯化方法,目前引物合成如此便宜,qPCR值得你进行PAGE及以上的纯化方式,而合成仪器这些信息并不重要,很多人做了几十年引物也不知道合成仪是ABI3900。
关于引物设计原则,大家大可不必死记硬背,因为大多数引物设计软件或者在线工具都能够兼顾这些问题(推荐在线工具
http://primer3.ut.ee/
),再说99.999%的引物设计都不是靠人工去看的,笔者有时候一天设计上百条引物,如果一条条看,那不得变成斗鸡眼了。
只需要在引物设计好以后,检查一下以下几点:
1、靠近3’端设计引物:对于用oligo dT引物进行cDNA第一链合成的情况,考虑到反转录效率问题以及RNA完整性问题,设计引物需要靠近3’端设计,以提高扩增效率。用一个图解释如下(这都看不懂就没有办法了):
为什么要靠近3’端设计引物
2、TM值:Tm值在55-65℃(因为60℃核酸外切酶活性最高),GC含量在40%-60% 。
3、BLAST:为了避免对基因组的非特异性扩增,必须用Blast去做补充验证。
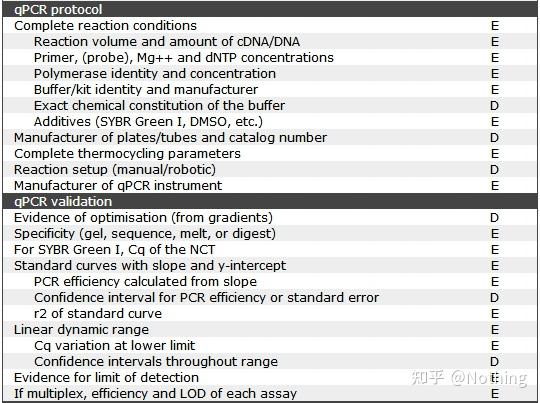
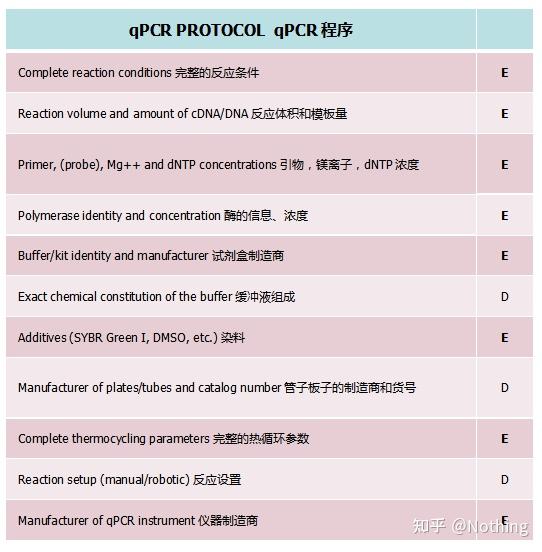
MIQE之荧光定量PCR过程和程序
1.qPCR试剂盒
按照MIQE要求,我们必须在文章中清楚的描述完整的反应条件,包括PCR的反应体系配置,用的什么试剂盒,制造商是谁,多大的反应体系,用的是染料法还是探针法。事情并非仅仅描述那么简单,这就牵涉到大家要如何选择荧光定量试剂盒。
目前,荧光定量PCR试剂盒的制造生产已经是一个非常成熟的技术,只要不是选择极偏极烂的厂商,试剂有问题基本上是一个中500万一样的概率,不过我们还是要跟大家普及几点:
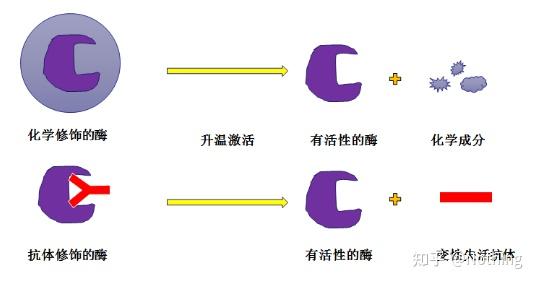
热启动Taq酶
PCR最重要的部分是热启动Taq酶,市面上的热启动酶一般分为两种类型,一种是化学修饰的热启动酶(你可以想象成石蜡包埋),一种是抗体修饰的热启动酶(抗原抗体结合)。化学修饰是早期的热启动酶存在方式,达到一定的温度,酶就释放活性。抗体修饰的热启动酶是用生物学的办法封闭酶的活性,当到达一定温度,抗体作为蛋白就变性失活,酶的活性就发挥出来了。
然而,这个有什么用呢?是这样,抗体修饰的酶释放活性比化学修饰的酶释放活性要快,所以在灵敏度方面,抗体修饰的酶略占优势,以至于市面上的试剂盒基本上已经没有化学修饰的酶了。如果有,那么这个厂家还技术还停留在千禧年的时代。
镁离子浓度
镁离子浓度在PCR反应中至关重要,合适的镁离子浓度能够促进Taq酶活性释放,浓度过低,会显著降低酶活性;浓度过高又使酶催化非特异性扩增增强。镁离子浓度还会影响引物的退火、模板与PCR产物的解链温度,从而影响扩增片段的产率。
镁离子离子的浓度一般控制在25mM,当然了,一个好的试剂盒,镁离子浓度一定是控制的比较好的。有的商家在试剂里面加入镁离子的螯合剂,可以达到镁离子浓度自动调节的作用。
荧光染料浓度
荧光染料,也就是我们通常用的SYBR Green,该染料主要是靠结合在双联DNA的小沟中发生荧光,由于该染料对双链DNA的结合是非特异性的,也就是说只要是双链DNA与其结合,都能发生荧光,所以体系中的引物二聚体、DNA模板等都会与其结合,形成本底信号。由于其光敏的特性,市面上的产品一般都用棕色离心管包装(如下图)。
荧光染料的浓度也很重要,浓度过低导扩增曲线后期上不去,不完美;浓度过高会造成噪点干扰。由于荧光定量PCR主要是看CT值,所以如果调不好荧光染料的浓度,低点比高点好。当然合适的染料浓度是最好的。
ROX
ROX染料是用以校正孔与孔之间产生的荧光信号误差。某些仪器厂家需要校正,某些不需要。例如使用Thermo Fisher Scientific公司的Real Time PCR扩增仪通常就需要校正,包括7300、7500、7500Fast、StepOnePlus等。一般试剂盒说明书都会有所描述。
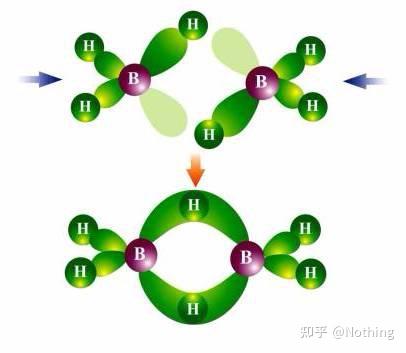
弱氢键处理
弱氢键的处理是一个比较有技术含量的事情,小编翻看了很多试剂盒的说明书,都没有提到过这个话题,
其实它是那么的重要。碱基的结合主要靠氢键的力量,强氢键就是正常扩增,弱氢键导致非特异性扩增,如果不能很好的消除弱氢键,非特异性扩增就无法避免。
在笔者目及范围内,只有少数几家公司注意到这个问题。各位在购买试剂盒的时候可以对照参考你要选择试剂盒是否在这方面考虑过解决方案。
反应体积
20-50ul体系是比较常用的,更小的体积容易造成误差,一般来说,试剂盒的说明书会有推荐使用PCR反应体积,大家切勿自作聪明,用更小的体积以达到节省成本的目的。商家推荐使用体积,其实是经过测试的,也可能就是他们无法解决小体积造成误差这个问题。
2.管子板子的制造商和货号
荧光定量PCR的原理大家都知道,荧光收集主要是通过PCR管盖进行的,选择PCR耗材注意两点:透光性好,适配仪器。一般来说主流品牌的板子和管子都没问题,但是在适配方面得谨慎选择,要不然会上不了仪器。
3.完整的热循环参数
(不解释,哪个PCR不要呢?)
4.仪器制造商
(不解释,就那么几个品牌。)
<hr/>
4.顶阶认识
MIQE之qPCR验证
这是是qPCR的
重中之重!
多少英雄好汉都在这里折戟沉沙,当然也有可能你运气比较好,研究的基因也简单,所以顺着风飘过了冰窟窿。
qPCR的验证信息意在检验数据的可靠性,我们把必要的验证信息罗列如下:
1.特异性检验
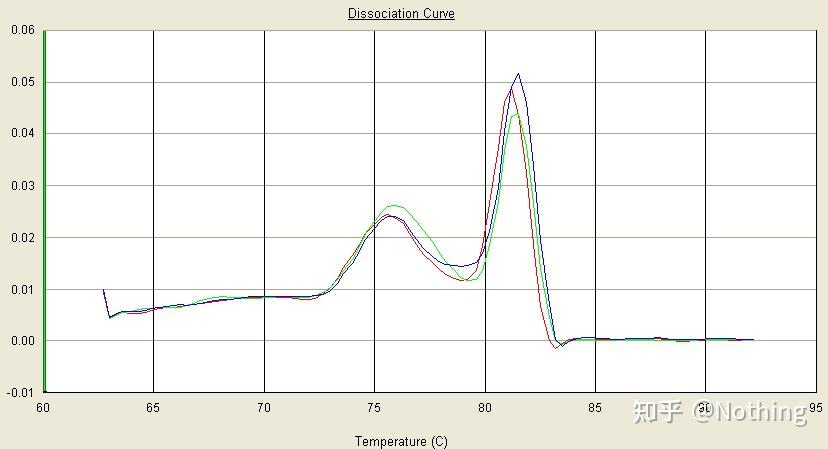
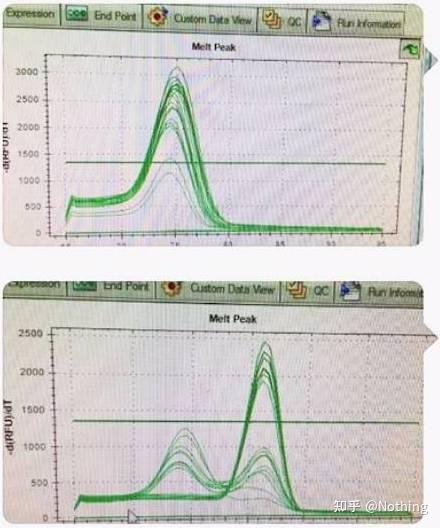
通过检测电泳图片是否是单一条带;测序验证;熔解曲线看峰图是否单一;酶切验证等方法,检验目的基因扩增的特异性。
在此,我们着重介绍
用熔解曲线的方法分析非特异性扩增
的问题,一般来说由于我们引物设计的时候要求产物片段大小在80-200bp这个范围,这使得PCR产物的熔解温度在80-85℃的范围。
所以如果有杂峰的情况肯定是有其他非特异性扩增产物出现;如果波峰出现在80℃以下,一般考虑是引物二聚体;如果波峰出现在85℃以上,一般考虑是有DNA污染或者更大片段的非特异性扩增。
注意!有时候只有一个80℃的单峰,这时候一定要坚持这个理念,很可能该扩增结果全部是引物二聚体。
正常的熔解曲线(单一峰无非特异性扩增)
有问题的熔解曲线(杂峰有非特异性扩增)
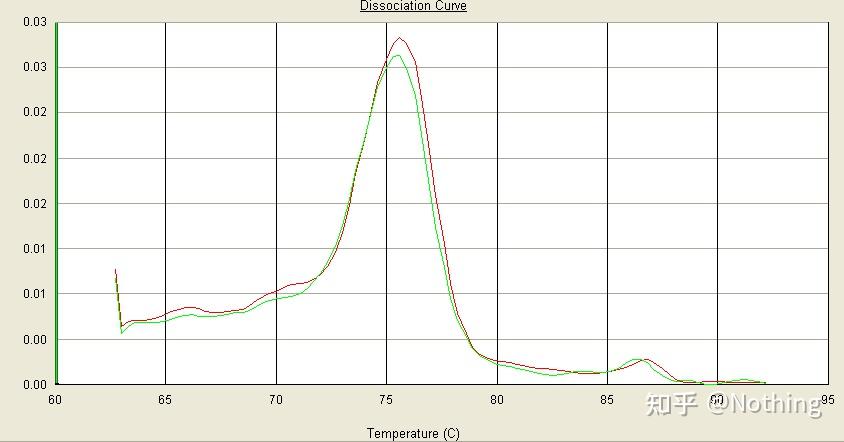
【案例分析】
有主峰,但是引物二聚体严重
下图这个单峰的溶解曲线很容易欺骗你的眼睛,认为是很完美的实验,结果完全错了,这时候我们得看溶解温度,波峰温度在80℃以下,完全是引物二聚体。
无目标片段,全是引物二聚体
在此,哥哥有点欲罢不能了,下图是一个学渣发给我的用手机拍的照片,他使用的试剂都是业界常用的品牌,从一个T字头品牌更换为另外一个T字头品牌,我想你们已经猜出来了。学渣向我哭诉:“第一个图片使用的试剂太好了,波峰单一,后来使用你推荐的试剂以后,变成第二个图的样子,出现了杂峰。你把我坑惨了。”
窦娥冤,事实上,我们很容易被这种图片麻痹,仔细分析后发现:第一个图波峰处于75℃,完全是引物二聚体;第二个图波峰分别出现在75℃和82℃,至少还有产物出现。
学渣反馈的图片
所以根本的问题不是试剂问题,而是引物设计的问题,同时也证实了某些大牌并不是铁打的质量,也应证了哥哥之前说的那句话:不是试剂品牌撑起了你的文章,而是你的文章撑起了试剂品牌。试想,如果这个学渣没有更换试剂,这个错误的数据就这样拿到水刊去,等来的也是悲剧。
2.空白对照的Ct值
不解释,如果空白对照有Ct值,不就是污染了吗?
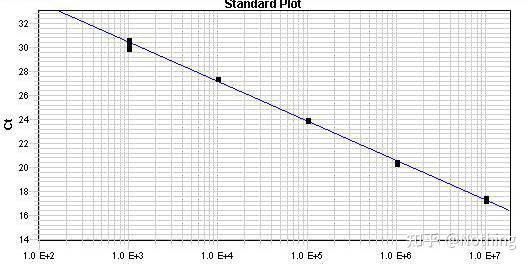
3.标准曲线
包括斜率和计算公式,通过公式可以计算PCR效率,一个完美的实验,要求
标准曲线的斜率slope趋近于3.32,R² 趋近于0.9999。
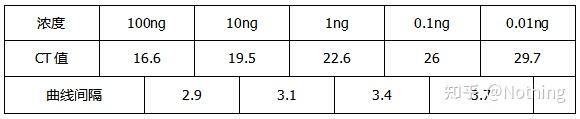
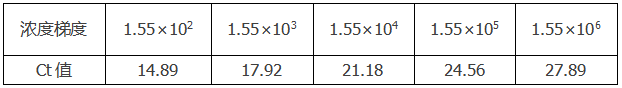
举个栗子:我们按照下表的浓度梯度得出标准曲线和扩增效率。
4.线性动态范围
反应的动态范围是线性的,根据生成标准曲线的模板,动态范围应当包括至少3个数量级的梯度,理想状态5个浓度梯度,并且注意高浓度梯度和低浓度梯度下Ct值的变化。
5.检测精度
qPCR结果变化,也就是重复性不好,也即精度差的问题,是由很多因素导致的,包括温度、浓度、操作等都会影响。qPCR精度的一般随着拷贝数减少而变得更不可控。
理想的状态是实验内变化,这种技术上的变化应该有别于生物变异,生物复制可直接解决不同组或治疗组之间的qPCR结果的统计学差异。特别是诊断分析,必须报道不同部位和不同操作者之最批间精度(重复性)。
6.检测效率和LOD(多重qPCR中)
LOD为检测到阳性样本最低浓度为95%。换句话说,包含一组靶基因复制内LOD的浓度最多不超过5%失败反应。
在做多重qPCR分析时,特别是就应用于同时检测点突变或多态性检测,多重qPCR需要提供证据表明多个目标片段的准确度在同一管中不受损,多重检测和单管检测的效率和LOD应该是相同的。特别是高浓度靶基因和低浓度靶基因同时扩增时,必须注意这个问题。
问题和解决方案
一般来说qPCR调试经常遇到的问题集中在以下几个方面:
■ 出现非特异性扩增
■ 引物浓度难以选择和引物二聚体的困扰
■ 退火温度把握不准
■ 二级结构影响扩增效率
非特异性扩增
出现
非特异性扩增
,一般想到引物设计是不是不太合适,但是在不急着更换引物的情况下,可以先试试以下办法(原理也附上):
• 提高退火温度——尽量让弱氢键无法维持
• 缩短退火、延伸时间——减少弱氢键的结合机会
• 降低引物浓度——降低多余引物和非目标区域的结合机会
与非特异性扩增相反的情况——
扩增效率低
,应对扩增效率低的措施也刚好相反:
• 延长退火、延伸时间
• 改为三步PCR,降低退火温度
• 提高引物浓度
Ps:
很多90后研究生都不愿意再去研究如何调试实验,希望试剂盒就能够完全解决问题(如果毕业后想去试剂公司做研发的另说),实际上试剂厂商也是这样思考的,希望傻瓜拿到都会用,所以试剂厂商在解决非特异性扩增的问题上花了不少功夫,包括引入弱H键吸收因子。
要想轻松解决问题,傻瓜们还是要会看试剂公司的介绍,有没有吸收弱氢键的因子。
引物浓度难以选择和引物二聚体的困扰
法门1
:一般来说qPCR的试剂盒说明书是有推荐体系和推荐引物浓度的。
法门2
:通过设置引物浓度梯度的方法进行调试。
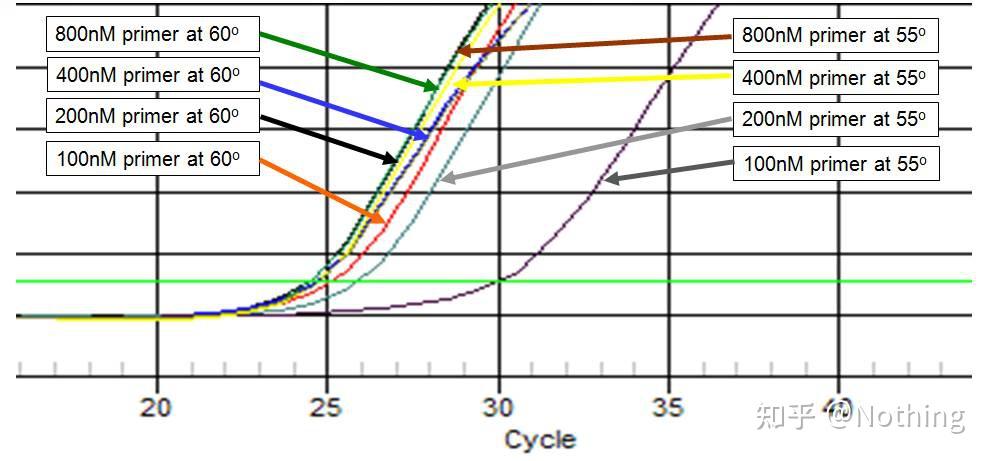
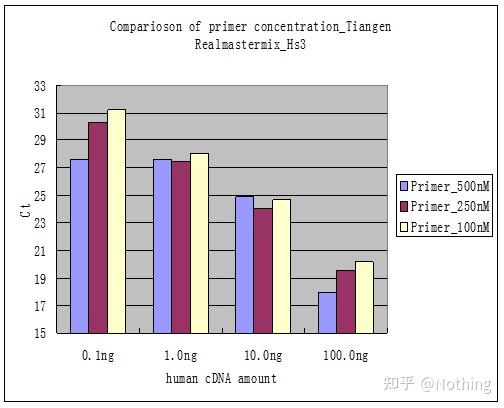
下面盗用某公司图片来做个说明,下图是用三个引物浓度梯度(100nM、250nM、500nM)和四个模板浓度梯度(0.1ng、1ng、10ng、100ng)做出来的荧光定量结果,根据实验结果的Ct值作图如下:
引物浓度选择
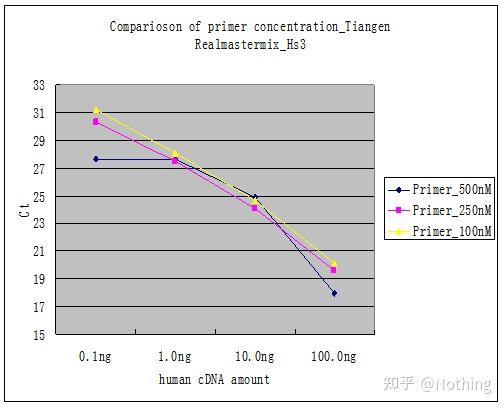
把每个引物浓度连成一条线如下:
引物浓度选择
很明显,100nM、250nM的引物浓度线性关系比较好,500nM的引物浓度线性关系比较差。而100nM、250nM中,250nM的Ct值比较小,所以最佳引物浓度是250nM。
一般来说严重的引物二聚体在熔解曲线可以看到。如果设计的引物避免不了引物二聚体怎么办?
法门3
:减少引物用量,升高退火温度(不用解释)。
退火温度的经验值是60℃。如果把握不准,怎么筛选比较合适的退火温度呢?答案跟引物浓度的选择是一样的——
梯度测试
。
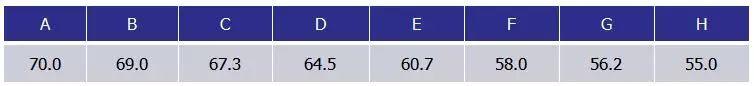
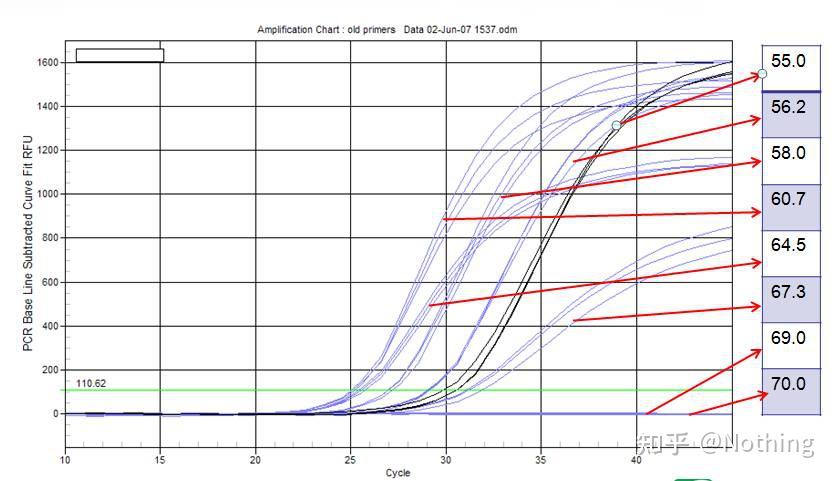
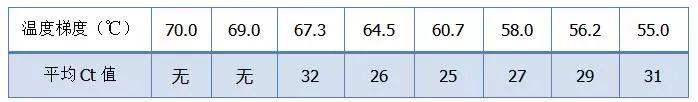
拿个Bio-rad公司的图来说明问题,对某一个目的片段的扩增,设置八个温度梯度,每个做三个重复,得到的扩增曲线如下:
退火温度选择
解读:
◥ 70℃、69℃——基本上引物都无法结合,所以没有扩增。
◥ 67.3℃——开始有少量扩增,Ct值比较大。
◥ 64.5℃——Ct值减小。
60.7℃、58.0℃、56.2℃、55.0℃这几个温度下Ct值基本上趋于稳定,只是最后的荧光值有区别。又该如何选择呢?
原则:Ct值靠前是第一原则,同样的Ct值选择较高的退火温度,以避免二聚体和非特异性扩增。55℃虽然有较高的荧光值,但是里面可能会有二聚体或者非特异性扩增。
二级结构影响扩增效率
再拿Bio-rad公司的图片来说明问题,同样是设计温度梯度来扩增一个含有二级结构的基因。
二级结构出现
可以看出,随着温度梯度的下降,开始有产物出现并且Ct值往前靠,在60.7℃达到最小值,之后随着温度梯度下降,Ct值变大。反过来说,随着温度提高,二级结构打开,扩增效率上升,当到达一定温度后,再升高温度也无法提高扩增效率。因为这个时候引物都无法稳定结合了。
所以,寻找Ct值最低的温度,这个温度就是扩增二级结构模板的最佳温度! 当然聪明的傻瓜们一定知道,非必须的情况下,最好更换引物,避开二级结构区域。
值得收藏!!!
原文地址:https://zhuanlan.zhihu.com/p/349018075
回复
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2026
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X5.0 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-5-10 05:31
发表于 2025-5-10 05:31