SARS-CoV-2是一种正链RNA病毒,它的复制是由病毒非结构蛋白(nsp)的多亚基复制/转录复合物介导的,这种复合物的核心成分是RNA依赖性RNA聚合酶(RdRp)的催化亚基(nsp12)。nsp12本身几乎没有什么活性,其功能需要包括nsp7和nsp8在内的辅助因子,这些辅助因子可以增加RdRp的模板结合和持续合成能力。 RdRp也被提出是一类称为核苷酸类似物的抗病毒药物---包括瑞德西韦(remdesivir, 也称为GS-5734)---的靶点,其中瑞德西韦是一种前体药物,在细胞内可转化为三磷酸形式的活性药物。
RdRp一直是结构生物学研究的重点 科学家们已解析出nsp7、nsp8以及nsp12-nsp7-nsp8复合物的结构,并提供了RdRp复合物的整体结构。然而,由于没有SARS-CoV-2 RdRp与RNA模板或核苷酸抑制剂所形成的复合物的结构,药物发现工作受到阻碍。 相关研究结果近期发表在Science期刊上,论文标题为“Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir”。
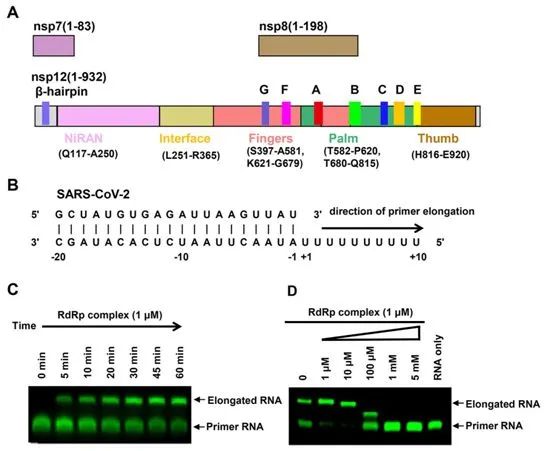
图1.nsp12-nsp-7-nsp8 RdRp活性复合物的组装以及瑞德西韦对它的抑制作用 纯化的nsp12在与长50个碱基的部分双链模板-引物RNA的结合上表现出很小的活性(图1B),这与SARS-CoV的nsp12相类似。nsp7和nsp8的存在显著增加了nsp12与模板-引物RNA的结合。在加入三磷酸腺苷(ATP)后,nsp12-nsp7-nsp8复合物在聚尿嘧啶模板上也表现出RNA聚合活性(图1,B和C)。这种RNA聚合活性可被加入的瑞德西韦的活性三磷酸形式(triphosphate form of Remdesivir, RTP)有效抑制(图1D)。即使在10 mM ATP的存在下,1 mM RTP也完全抑制了RdRp的聚合活性。
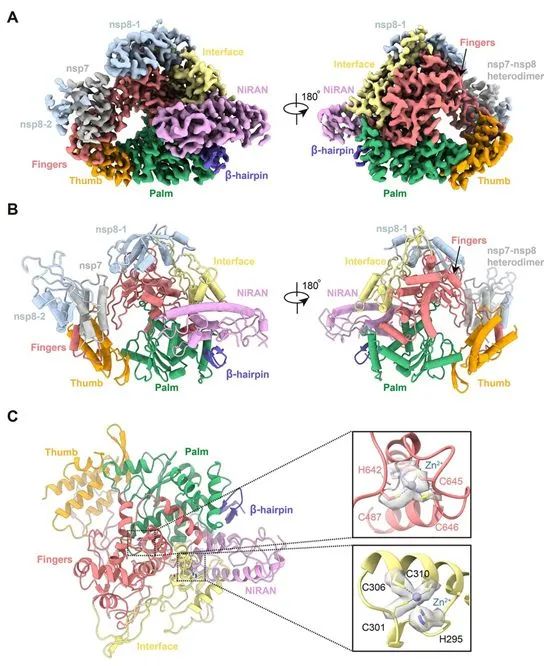
纯化的RdRp复合物在53℃的解链温度下相对稳定 相比之下,作为一种前体药物,瑞德西韦在5 mM浓度下,对这种纯化的RdRp酶的聚合活性没有任何抑制作用,瑞德西韦的单磷酸形式(Remdesivir in its monophosphate form, RMP)也没有这种抑制能力。 纯化的RdRp复合物在53℃的解链温度下相对稳定。nsp12-nsp7-nsp8复合物的阴性染色电镜可视化观察显示出具有良好均匀性的分散颗粒。对于apo形式下的nsp12-nsp7-nsp8复合物,这些研究人员在洗涤剂DDM的存在下,对这种复合物样本进行了玻璃化处理。 对图像处理的初步尝试显示,这些颗粒是优先取向的。因此,他们收集了超过570万个颗粒的7400多个显微影像,以增加非优先取向的投影数量。其中,81494个颗粒被用于产生2.8埃分辨率的密度图。 首先,大多数颗粒被吸附到低温电镜网栅条上,而不是停留在玻璃态冰中。其次,在低温电镜样本制备的条件下,RNA双链可能从template-RTP RdRp复合物中解离下来。 最终,这些研究人员制备出15mg/ml的template-RTP RdRp复合物样本用于低温电镜实验,这一浓度远高于可溶性蛋白复合物在低温电镜研究时所使用的正常浓度,这种template-RTP RdRp复合物的高浓度具有质量作用效应,以稳定这种RNA-蛋白复合物,并且让过量的template-RTP RdRp复合物逃避低温电镜网栅条的吸收,从而进入玻璃态冰中。 他们收集了2886个显微影像,从而利用130386个颗粒投影产生了2.5埃分辨率的结构。由于这种解析出的结构的相对较高的分辨率,这种低温电镜图清晰地显示了这种复合物的关键结构特征。 在NiRAN结构域之后是一个由3个螺旋和5个β链组成的界面结构域(残基251-365),该界面结构域与RdRp结构域(残基366-920)相连(图1A和2B)。nsp12的RdRp结构域显示出典型的杯状右手构象,手指亚结构域(残基397-581和残基621-679)与拇指亚结构域(残基819-920)形成一个封闭的环形结构(图2,A和B)。这种封闭构象通过nsp7和nsp8的结合而变得稳定,其中一个nsp8分子位于手指亚结构域的顶部,并与界面结构域相互作用。 nsp12的封闭构象可通过nsp7-nsp8异源二聚体进一步稳定化,这种异源二聚体沿着拇指-手指亚结构域界面堆积(图2,A和B),此外,这些研究人员能够在由H295-C301-C306-C310和C487-H642-C645-C646组成的保守性金属结合基序中配位两个锌离子(图2C),这一点在SARS-CoV RdRp结构中也能观察到, 这些锌离子很可能是维护RdRp结构完整性的保守结构成分。
图2.apo形式下的nsp12-nsp-7-nsp8 RdRp复合物的低温电镜结构。
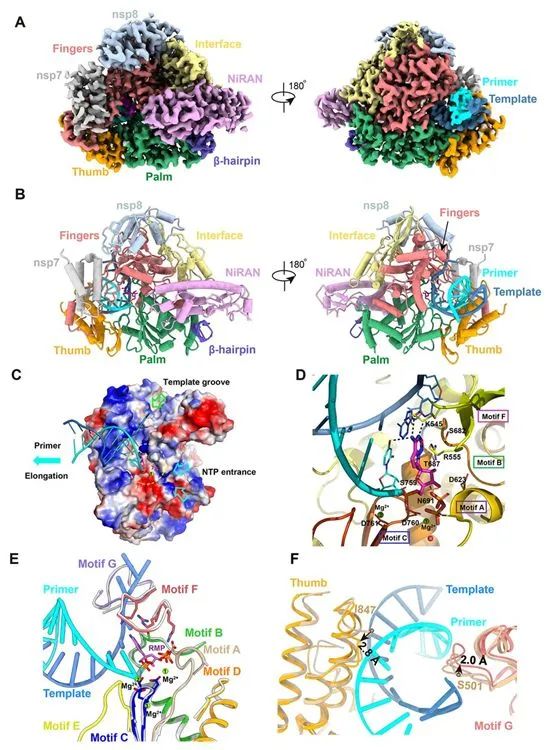
图3.RdRp复合物与瑞德西韦与RNA结合在一起时的低温电镜结构。
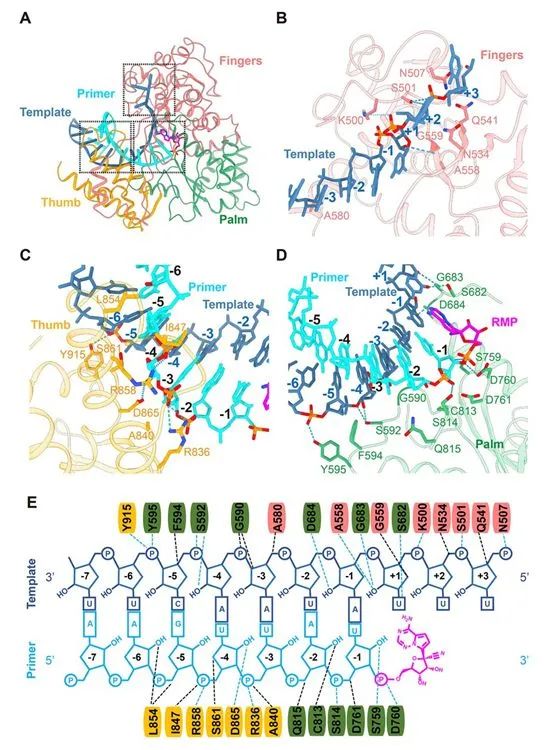
通过非专一性RNA链终止机制抑制病毒RdRp活性 令人惊讶的是,尽管nsp7和nsp8这两种蛋白是RdRp结合RNA所需要的,但是它们并不介导RNA相互作用。大多数蛋白-RNA相互作用涉及RNA磷酸-核糖骨架,许多相互作用直接发生在2′-OH基团上(图4E),从而为区分RNA与DNA提供了基础。nsp12与模板-引物RNA的任何碱基对都没有接触,这表明序列独立于DNA的生物碱基对。 nsp12与模板-引物RNA的任何碱基对都没有接触,这表明RdRp与RNA的结合与序列无关,这与RdRp在延伸阶段的酶活性不需要特定的序列这一事实是一致的。 因此,瑞德西韦和许多核苷酸类似物前体药物一样,通过非专一性RNA链终止机制抑制病毒RdRp活性,这一机制需要瑞德西韦转化为它的活性三磷酸形式。
图4.RdRp复合体识别RNA。
RMP还与K545和R555的侧链发生相互作用,在结合的RMP附近有两个镁离子和一个焦磷酸。这两个镁离子与磷酸二酯骨架相互作用,它们是催化活性位点的一部分。焦磷酸位于活性位点的核苷酸进入通道的通道口,可能阻断核苷酸三磷酸进入活性侧(图3,C和D)。 基序F中与+1碱基相接触的K545和R555侧链与引物链RNA相互作用(图3D),从而将进入的核苷酸稳定在正确的位置上进行催化。模板-引物RNA在活性位点中的的定位类似于模板-引物RNA在脊髓灰质炎病毒RdRp延伸复合物和HCV NS5B RdRp抑制复合物中的定位,参与RNA结合的残基以及包含催化活性位点的残基都是高度保守的,这突出了RdRp在这些不同的RNA病毒中的保守性基因组复制机制,并表明有可能开发出广谱抗病毒抑制剂,如瑞德西韦和加利德韦(galidesivir, 也称为BCX4430)。 第三,基序G残基K500和S501也向外移动2.0埃,以容纳模板链RNA的结合。除了这些变化之外,apo形式下的nsp12和template-RTP RdRp复合物中的nsp12非常相似:整个蛋白中所有Cα原子的均方根偏差(root mean square deviation, rmsd)为0.52埃。 特别是构成催化活性位点的结构元素可以完全叠加(图3E),这表明SARS-CoV-2 RdRp是一种相对稳定的酶,当与RNA模板结合后,就可以发挥复制酶的功能。病毒RdRp是一种高度进行性的酶,其复制速度可达100个核苷酸/秒。 apo形式下的结构和活性酶结构之间没有明显的构象变化,这与病毒RNA聚合酶的较高持续合成能力相一致,因而在复制周期中不需要消耗额外能量来导至活性位点发生构象变化。
RdRp是现有许多核苷酸类药物的主要靶点 除了瑞德西韦之外,一些核苷酸类似物药物,包括法匹拉韦(Favipavirir)、利巴韦林(Ribavirin)、加利德韦(galidesivir)和EIDD-2801,在细胞实验中有效地抑制SARS-CoV-2复制。 与瑞德西韦一样,这些核苷酸类似物也被提出通过非专一性RNA链终止机制抑制病毒RdRp,这种机制需要前体化合物转化为它们的三磷酸活性形式。template-RTP RdRp复合物的结构提供了一个很好的模型来合理地思考这些药物如何抑制SARS-CoV-2 RdRp活性。 特别是,EIDD-2801在阻断SARS-CoV-2复制方面比瑞德西韦强3~10倍。胞苷环外的N4羟基与K545的侧链形成一个额外的氢键,而胞苷碱基也与模板链上的鸟嘌呤碱基形成一个额外的氢键。这两个额外的氢键可能解释了EIDD-2801在抑制SARS-CoV-2复制方面具有明显更高的效力。 在病毒酶中,RdRp是现有许多核苷酸类药物的主要靶点。在这篇论文中,这些研究人员报道了SARS-CoV-2 RdRp复合物的apo形式以及与模板-引物RNA和活性形式的雷德西韦结合在一起时的结构。这些结构揭示了模板-引物RNA是如何被这种酶识别的,以及瑞德西韦如何抑制链的延伸。 结构比较和序列比对表明,RdRp识别底物RNA和瑞德西韦抑制RdRp的模式在不同的RNA病毒中高度保守,这为设计基于核苷酸类似物的广谱抗病毒药物提供了基础。 此外,这些结构为现有的核苷酸类药物(包括强效的EIDD-2801)的建模和修饰提供了一个坚实的模板。总之,这些观察结果为设计更强效的抑制剂来对抗SARS-CoV-2的恶性感染提供了合理的基础。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号