CRISPR/Cas9是一种强大的基因编辑工具,广泛应用于从基础研究到治疗多种人类疾病的领域,尤其在单基因遗传病的治疗中,有望实现一次性治疗、长期有效的效果。
然而,CRISPR/Cas9系统存在一个关键应用瓶颈:脱靶效应。这意味着它可能在基因组的非预定位置进行切割,这是由于系统可以容忍sgRNA与DNA靶点之间的多个错配或序列凸起(bulges)。这种非预期的编辑会带来安全风险,限制了其在基因治疗中的应用。
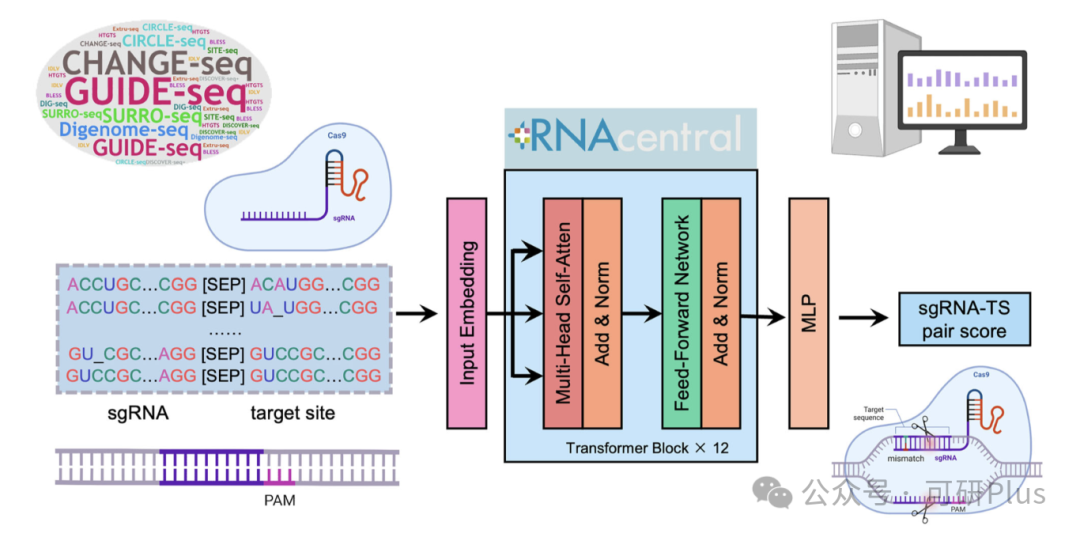
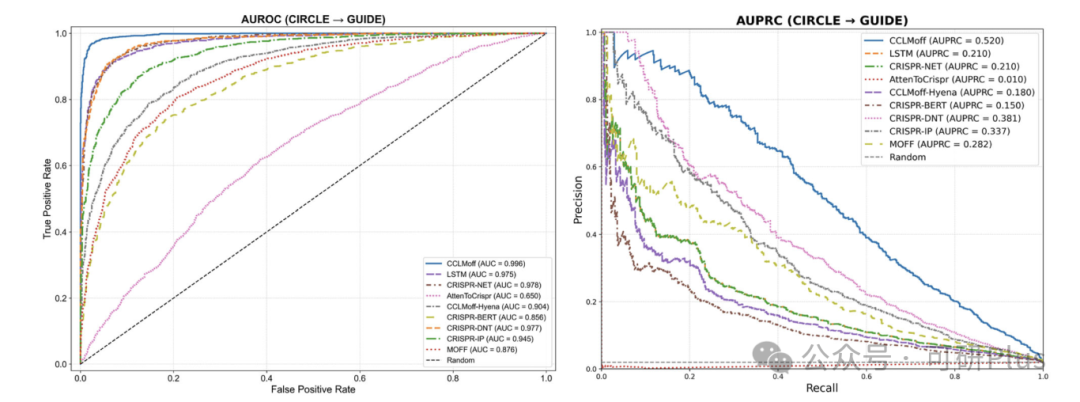
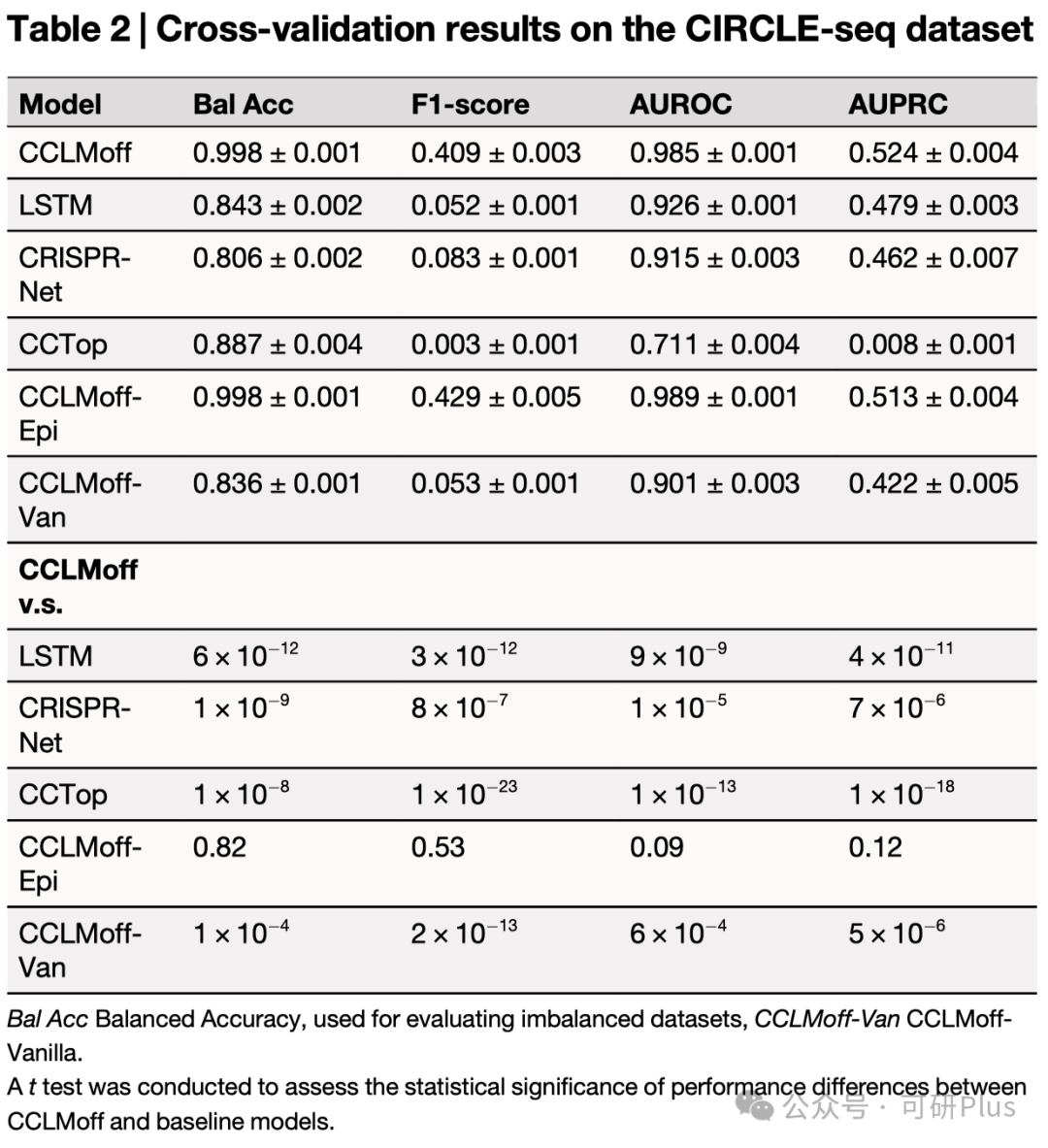
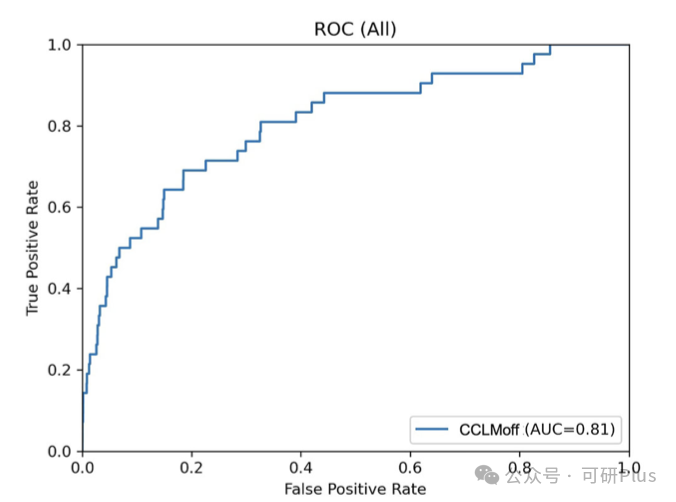
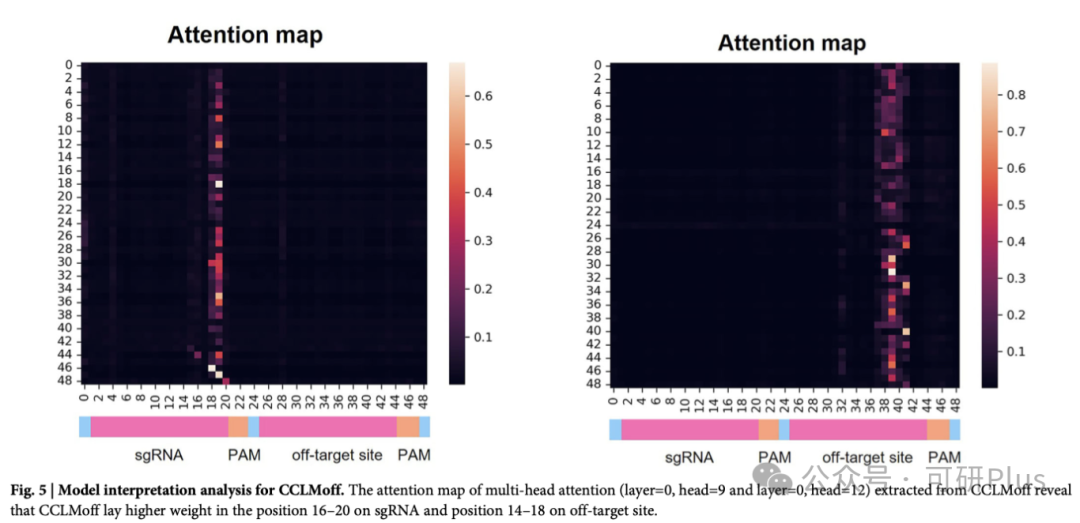
2025年6月6日,Communications Biology在线发表了佛山大学唐冬生、深圳市卫生健康发展研究和数据管理中心赵靓联合团队的最新研究进展:“A versatile CRISPR/Cas9 system off-target prediction tool using language model”。该研究建立了一个基于RNA语言模型的通用脱靶预测模型:CCLMoff,可以更准确地识别CRISPR/Cas9系统的脱靶位点。该工具利用RNA语言模型和全面的数据集进行训练,能够精准识别脱靶位点,并在不同实验条件下通用,克服了现有工具(如CRISPR-Net、Cas-OFFinder)泛化能力差的局限。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-6-11 12:08
发表于 2025-6-11 12:08


 提升卡
提升卡


