来源:小桔灯网 作者:oryzmeng 前言: 小编以产品设计开发的时间轴为主线,结合“人、机、料、法、环”梳理一张IVD注册质量管理体系自查表,希望可以对准备迎接现场检查的战友们有所帮助或启发。限于经验与阅历,不正之处欢迎交流与批评。
先介绍一个麦肯锡的分析法 MECE:“Mutually Exclusive & Collectively Exhaustive”,中文意思是“互相独立,完全穷尽”。MECE是结构化思维和表达的核心,能够不重不漏的将一个主题进行梳理和分解。
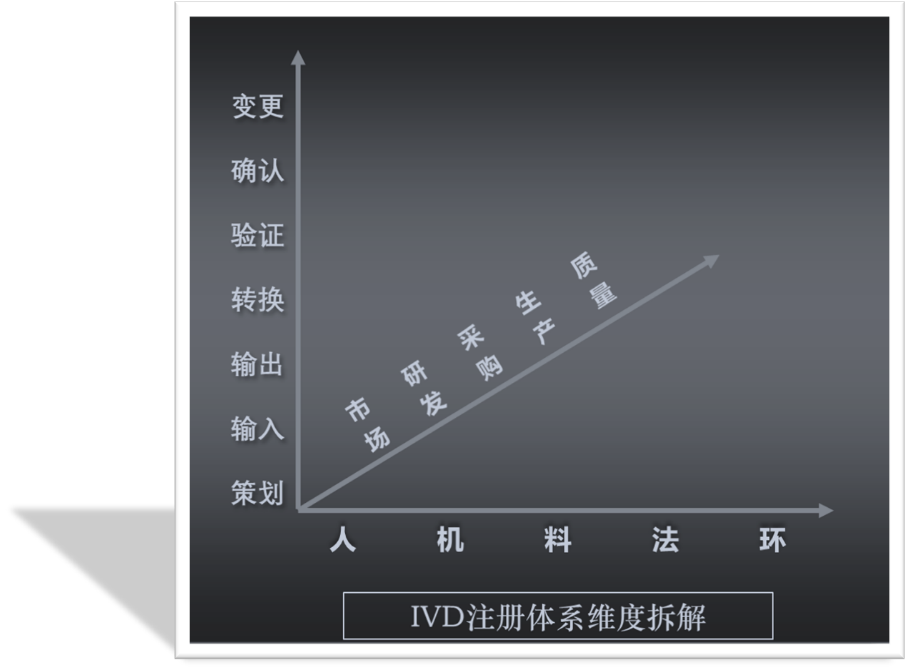
为了全面而又高效的梳理一个IVD产品从策划之初到设计开发输出、转换、验证、确认直至变更各个环节应开展的工作和产生的记录,我们借鉴MECE分析法,尝试以时间轴为主线保证互相独立,以“人、机、料、法、环”为支线完全穷尽,进一步的还可以分工到“市场、研发、采购、生产、质量……”等各个部门,从两维或三维角度来逐步的拆解和整理。
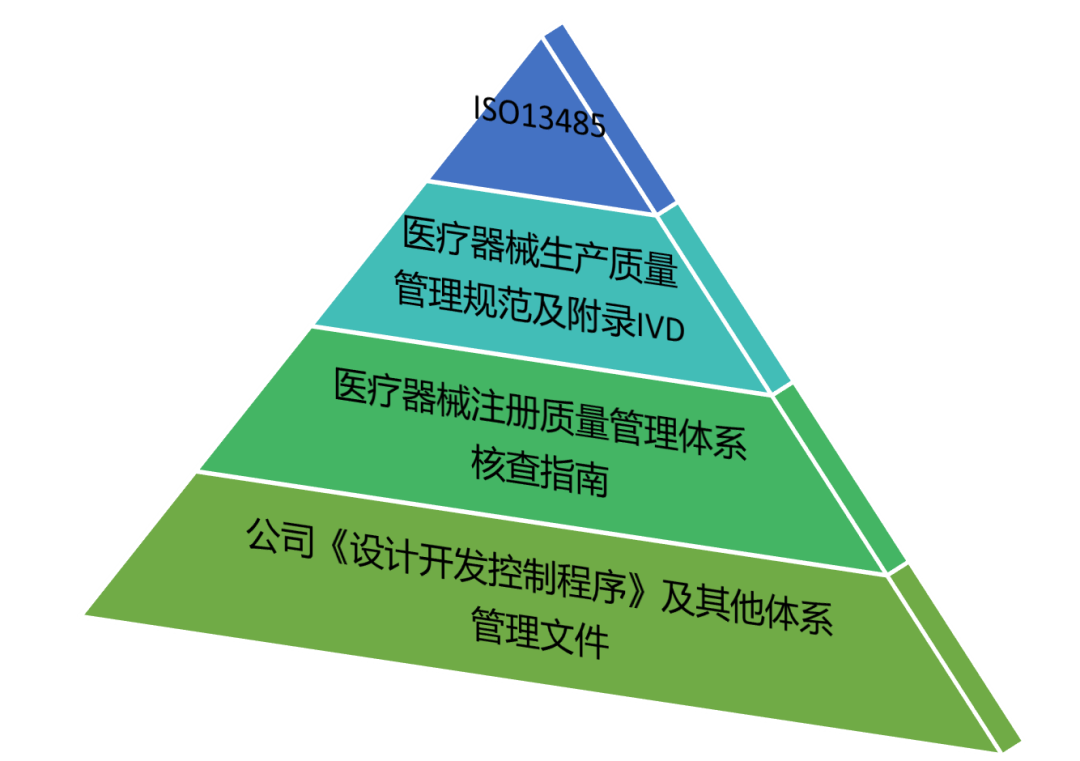
接下来准备下面四个文件及配套资料建立自查表: ✰ YY/T 0287-2017 医疗器械 质量管理体系 用于法规的要求(ISO13485:2016, IDT)是设计和开发要求的源头; ✰ 医疗器械生产质量管理规范及附录IVD是国内对ISO13485设计开发要求的转化(为便于应用建议直接使用医疗器械生产质量管理规范体外诊断试剂现场检查指导原则); ✰ 医疗器械注册质量管理体系核查指南对设计开发核查进行了细化; ✰ 公司自己的《设计开发控制程序》是最终的落地,正所谓灵魂要附体,就需要将上述的各种精神要求进行转化,直至可执行的层面。
举一个关于“输出”阶段不同法规要求的例子: 我们先用EXCEL表依次建立如下四列:
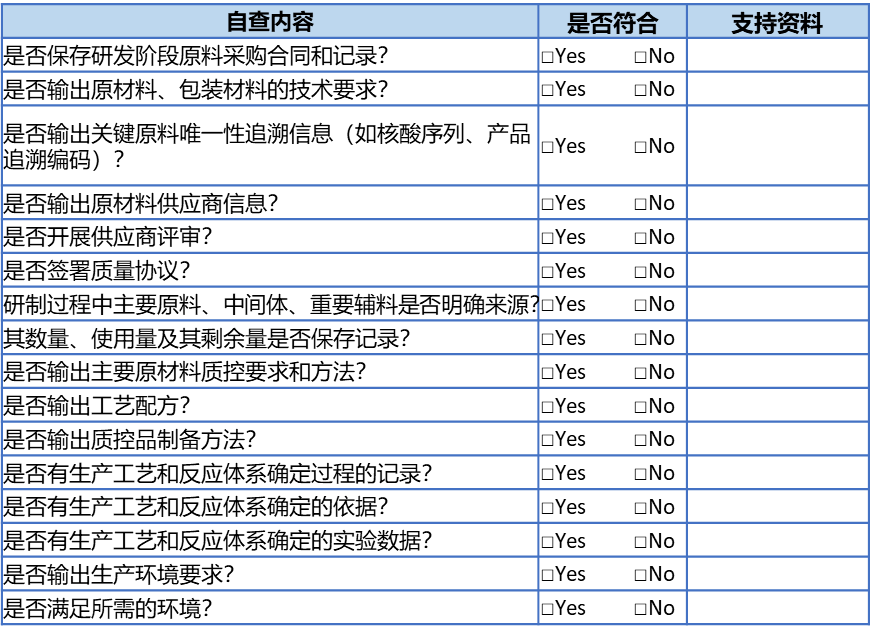
✰ ISO13485规定: 给出采购、生产和服务提供的信息; ✰ 医疗器械生产质量管理规范则细化为: 采购信息,如原材料、包装材料、组件和部件技术要求; 生产和服务所需的信息,如产品图纸(包括零部件图纸)、工艺配方、作业指导书、环境要求等; ✰ 医疗器械注册质量管理体系核查指南进一步要求: 体外诊断试剂应当重点关注原材料的供应商(生产商)信息,唯一性追溯信息(如克隆号、核酸序列、产品追溯编码等),主要原材料质量控制要求和方法,工艺配方,质控品制备方法及质量控制方法,以及所需设备仪器等。 核查生产工艺过程、反应体系如溶液配制、抗体包被过程、实验过程等的确定过程、确定依据、实验数据等。 综合上述要求,便可以针对该条制定如下自查内容:
单纯仅从法规的转化角度,这个表格(详细版本按下面链接自行下载)便可以使用了。然而每个公司对设计开发的阶段划分及所需输入和输出的要求都不尽相同,为了更适用于本公司应用,建议进一步结合《设计开发程序》进行细化和优化。
分享资料: 【1】注册体系核查自查表构建 素材分享版.xlsx 【2】YY/T 0287-2017 医疗器械 质量管理体系 用于法规的要求.pdf 【3】医疗器械生产质量管理规范体外诊断试剂现场检查指导原则.docx 【4】医疗器械注册质量管理体系核查指南.doc |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号