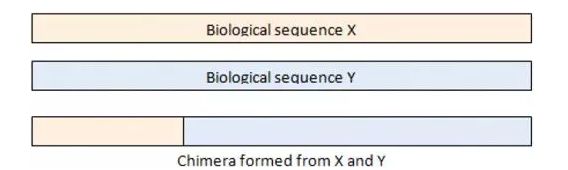
Q1:基于高通量测序的微生物多样性检测技术优势以及原理是什么? A:常规的微生物研究方法包括基因克隆文库、变性梯度凝胶电泳DGGE等,但这些方法的通病是信息量太小,且自然界中99%的微生物在实验室都没有办法纯化培养,不能充分反映复杂的环境微生物多样性和分布,且程序繁琐,效率低下,也无法检测到稀有菌群的种类,因此其重复性和分辨率都不甚理想。 Q2:微生物组测序设置生物学重复的意义是什么,一般情况下,设置多少个重复合适? A:生物学重复是指样本所有的环境因素、处理条件、采样时间都完全一致。生物学重复对于测序的实验设计以及后续信息分析都非常重要,设置生物学重复主要有以下几个作用: Q3:什么是嵌合体,嵌合体是否需要去除? A:嵌合体,遗传学上用以形容不同遗传性状的嵌合或混杂表现的个体。嵌合体序列是由来自两条或者多条模板链的序列组成,见下图:
图:嵌合体示例图 PCR反应中,在延伸阶段,由于不完全延伸,就会导至嵌合体序列的出现。在扩增序列X的过程中,序列延伸阶段,只产生了部分X序列延伸阶段就结束了,在下一轮PCR的反应中,这部分序列Y的引物接着延伸,扩增就会形成X和Y的嵌合体序列。在PCR过程中,大概有1%的几率会出现嵌合体序列,嵌合体在正常生物体中是不存在的,所以在16S扩增子测序的分析中,需要去除嵌合体序列。将去除后的cleandata再进行后续OTU聚类及分析。 Q4:Alpha多样性和Beta多样性在微生物检测中的意义? A:Alpha多样性是指一个特定环境或生态系统内的多样性,主要用来反映物种丰富度和均匀度以及测序深度。Beta多样性主要是为了研究不同样品或处理组之间的物种群落结构差异,是生物多样性的重要组成部分。Beta多样性与Alpha多样性差异在于alpha多样性只是用来描述单个样品的物种多样性,而beta多样性则是比较不同样品(生态系统)之间的多样性差异。Beta多样性与alpha多样性一起构成了总体多样性或一定环境群落的生物异质性。 Alpha多样性包括Chao1、Observed_species、Goods_coverage、Shannon、Simpson指数,综合考量态系统的丰富度与多样性,Beta多样性分析通常由计算环境样品间的距离矩阵开始,该矩阵包含任意两个样品间的距离,主要通过主坐标分析(Principal coordinates analysis,PCoA)、主成分分析(Principal component analysis,PCA)、多维尺度分析(Multidimensional scaling,MDS)、聚类分析(Clustering analysis)、相似性分析(Analysis of similarities,ANOSIM)等方法,对群落数据结构进行自然分解并通过对样本排序(Ordination),从而观察样本之间的差异。 Q5:Alpha多样性指数之间有什么区别吗? A:Alpha多样性包括Chao1、Observed_species、Goods_coverage、Shannon、Simpson等5个指数。其中Chao1,Observed_species指数主要关心样本的物种丰富度(Richness)信息;Goods _coverage反映样本的低丰度OTU覆盖情况;Simpson/Shannon主要综合体现物种的多样性(Diversity)和均匀度(Eveness)。其中Chao1和observed_species主要是估计群落中包含物种的数目;Goods _coverage是指各样品文库的覆盖率,其数值越高,则样本中序列没有被测出的概率越低,该指数实际反映了本次测序结果是否代表样本的真实情况;Shannon指数来源于信息熵,Shannon指数越大,表示不确定性大。不确定性越大,表示这个群落中未知的因素越多,也就是多样性高;Simpson数值范围在0-1之间,当群里只有一种物种的时候,此时Simpson值最小为0,同时也是我们直观理解的多样性最小。当物种种类无限多(丰富度最高),并且每个物种数目都一致(均匀度最高)的时候,Simpson值为最大为1。根据我们的计算公式可得:observed_species、chao1指数高,说明样品物种数目多;shannon、simpson指数高,说明物种丰度以及均匀度都很高。 Q6:如何查找感兴趣的物种信息及其丰度 A:所有样本中每个OTU对应的物种注释信息以及丰度信息(绝对丰度)文件路径 summary/6_taxonomy_community/OTU_table_with_taxonomy.xlsx
summary/6_taxonomy_community/*/1_abundance_stats/Group/*_abund_Group.xlsx Q7:16S测序物种注释常用的数据库有哪些?这些数据库的特点(或优缺点)是什么? A:16S测序物种常用的数据库有RDP, SILVA ,Greengenes等,其中SILVA 数据库由于更新比较及时,因此也是目前最常选用的参考数据库之一。Greengenes数据库相对于其它经常更新的数据库,劣势比较突出。 联川目前使用的注释库是RDP+NT-16S。RDP数据库是目前较常用的比对、注释的参考数据库,版本更新比较快,16S序列信息较全,但由于该数据库注释结果只到属水平,而部分客户还是想通过16S得到种水平注释信息,因此我们使用NT-16S(基于NT库提取整理的16S数据库)数据库做为种水平注释结果的补充,并以RDP数据注释结果为优先级。 Q8:16S物种注释中为什么会出现叶绿体? A:本身的叶绿体含量会比较高,而叶绿体和所研究16S扩增区段的序列相似性很高,也是能被扩增出来的。因此该从实验端用普通的引物是无法避免该问题的。 对叶绿体本身就很高的样本(如叶片等)给出如下建议: 1)预先处理减少叶绿体含量的提前处理,以减少后期测序结果里面的叶绿体序列; 2)使用去除叶绿体的特异性引物扩增,以减少可能产生的叶绿体序列。 Q9:16S测序与其他项目关联原理及注意要点? A:众所周知,16S rDNA 测序主要用于研究某一特定环境中微生物结构组成,及其在不同处理条件下微生物种类及丰度差异,主要优势在物种多样性层面。由于PICRUSt等分析软件也可以得到功能上的注释结果,但功能预测的结果只能作为参考。如果想对基因层面对微生物代谢功能更深入的研究,就需要结合其他研究手段(宏基因组,宏转录组等)研究复杂微生物群落变化,基因功能层面差异及挖掘潜在的新基因。 除了与宏基因组和宏转录组联合分析外,16S和代谢组学联合分析也是近年来研究的热点。由于微生物群落的复杂性,研究微生物如何通过功能改变对宿主产生的影响是很困难的。通过测定微生物代谢物发生的改变来研究菌群功能状态是一种有效的方法。通过一定的数据筛选规则,找到可能相关的菌群临床指标以及代谢物。如加上做一些单菌实验以及群体验证,前期猜想将更加好地得到证实。 Q10:测序前通过一代测序鉴定了菌株的信息没有问题,一定能判断菌的纯化就完全没有问题吗? |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号