上期微课堂,我们介绍了伴随诊断分类情况(微课堂 | 伴随诊断之分类知多少)。本期,我们重点了解一下欧美及中国的伴随诊断监管现状。 美国
美国是最早提出伴随诊断试剂概念并对其进行针对性监管的国家,并在伴随诊断试剂监管发展的早期扮演引领者的角色。FDA于2014年和2016年先后发布《体外伴随诊断试剂指导原则》《体外伴随诊断试剂与治疗药物联合开发指导原则草案》。两份指导原则对“个体化药物-伴随诊断试剂”联合研发方案的设计、执行和评估等方面提出了建议,旨在促进与加快联合开发进程。 日本及欧盟 2013年,日本和欧盟继美国之后也引入伴随诊断试剂定义,并正在积极制定针对性监管政策。2013年,日本PMDA发布《伴随诊断试剂与药物申请审批通知》,通知的内容包括伴随诊断试剂的定义以及“个体化药物-伴随诊断试剂”联合研发的注意事项等。欧盟从2013年议会对伴随诊断试剂定义的修正案到2015年欧盟新版体外诊断试剂法规里面对伴随诊断试剂的定义,欧盟的监管政策不断进行调整,调整后欧盟与美国关于伴随诊断试剂的监管政策比较相近。 中国
相比美国、欧盟和日本,目前我国尚未引入伴随诊断试剂定义,国内现阶段暂时没有系统性的伴随诊断监管法规或是指南,在我国仍按照体外诊断试剂进行注册管理。CFDA于2014年7月30 发布了《体外诊断试剂注册管理办法》(以下简称《办法》)。《办法》中对体外诊断试剂的产品分类、产品标准、产品研制、临床试验和注册申请、注册检测、变更注册、重新注册等相关问题一一做出明确规定并在2017年1 月25 日由局令第30号进行了修正。 诊断试剂产品注册的基本要求如下图所示:
在注册过程中,监管部门的评审主要关注以下几个方面:
从上可看出在体外诊断实际生产企业提出注册申请前,必须对产品进入深入研究和系统评价,并对批准后产品的安全性、有效性负责。
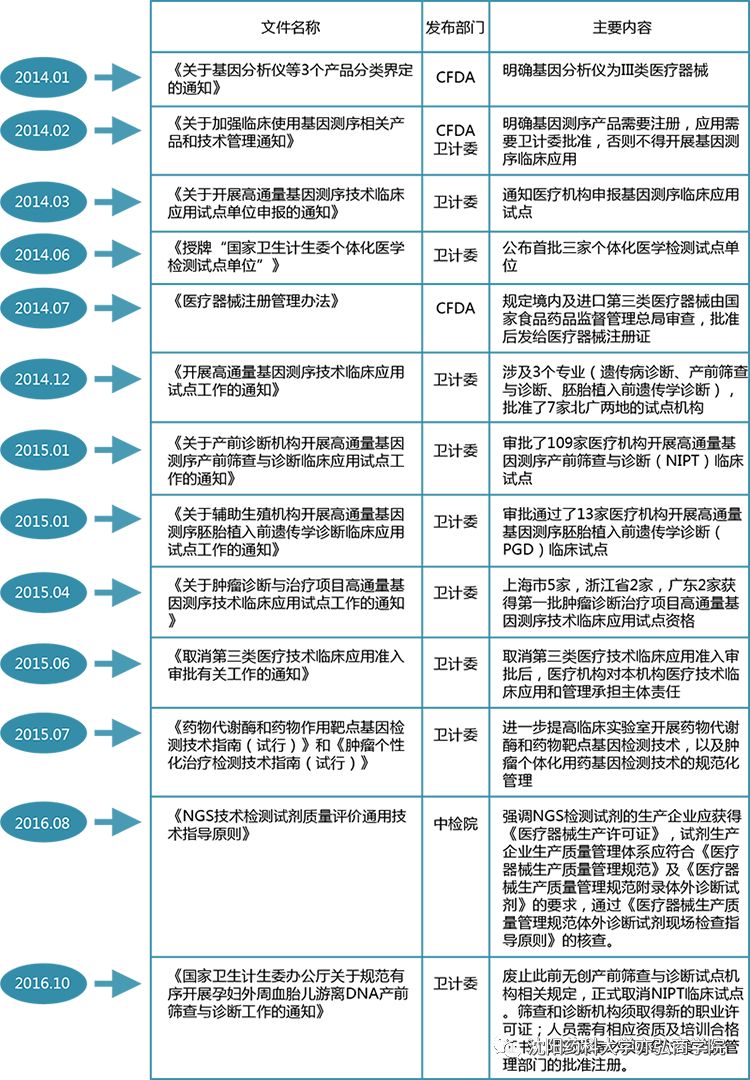
《办法》中根据产品风险程度由低到高,将体外诊断试剂分为第一类、第二类、第三类产品进行分级管理。境内第一类体外诊断试剂由设区的市级药品监督管理机构审查;第二类由省、自治区、直辖市药品监督管理部门审查;第三类由CFDA来确定类别分类。 CFDA在2017年发布《总局关于过敏原类、流式细胞仪配套用、免疫组化和原位杂交类体外诊断试剂产品属性及类别调整的通告》,调整了过敏原类、流式细胞仪配套用、免疫组化和原位杂交类体外诊断试剂的属性界定和分类原则,制定了产品分类列表并明确了有关实施要求。 关于近些年大热的新一代测序(NGS)检测技术,2014年以前,我国NGS处于无监管状态,2014年2月,CFDA和国家卫生和计划生育委员会(NHFPC)叫停了所有的NGS业务,对行业进行集中整顿,推出了一系列的NGS监管政策:
由于NGS涉及到的产业众多,所以监管部门也比较多,其中监督管理工作由CFDA主导,所有的NGS产品都需经CFDA注册,卫计委主要是对开展基因测序机构的资质进行审查和规范。 好啦,今天的微课堂就到这里呢~想要学习更多关于新药研发与伴随诊断同步开发的知识吗~欢迎来亦弘课堂学习讨论! 以上内容来源: 1. 学院2017年3月抗肿瘤课堂; 2. 卫计委临检中心副主任李金明:我国NGS诊断检测的监管之路 3. 体外诊断试剂注册管理办法(2017 版) |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号